
Abstract
Raltegravir (RAL) resistance is associated with the selection of integrase mutations at positions 92, 143, 148, and/or 155. A substantial proportion of RAL failures, however, occurs in the absence of these changes. An examination of RAL plasma concentrations may help in interpreting this observation. All early RAL virological failures seen at 22 clinics in Spain during 2009 were identified. HIV integrase sequences and RAL plasma trough concentrations (C t) were examined. A total of 106 patients experiencing virological failure on RAL were identified. Only the earliest sample on failure was examined. Integrase sequences could be obtained for 89 (84%), of whom 30 (33.7%) depicted primary RAL resistance mutations (15 N155H, eight Q148H/R, three Y143R, one E92Q, and three more than one of them). Another nine (10.1%) patients showed only secondary changes. The remaining 50 RAL early failures (56.2%) did not select any integrase change. RAL C t could be measured in 66 patients at failure and in 21 of them before failure. In a control group of 37 patients with viral suppression on RAL, detectable plasma levels were seen in all cases, with greater median RAL C t than in failures, either at the time of viral rebound (p<0.001) or before it (p=0.055). Moreover, median C t at the time of failure was greater in patients selecting primary RAL resistance mutations than in the rest of the failures (p<0.001). Undetectable RAL C t was seen only in patients failing RAL without integrase resistance mutations (64.1% of them). RAL failures in the absence of integrase resistance mutations mainly reflect poor drug compliance.
Introduction
The advent of highly active antiretroviral therapy (HAART) has dramatically improved the life expectancy of HIV-infected individuals, with estimates of gained survival increasing from 10.5 years in 1996 to 22.5 years in 2005. 1 However, HIV infection has no cure, and accordingly antiretroviral therapy must be given indefinitely in almost all patients. Problems in maintaining satisfactory drug adherence and overcoming side effects, along with the high rate of mutability and turnover of the virus, indicate that selection of drug resistance is a major concern over time. In this scenario, new antiretroviral drugs targeting different enzymes in the HIV replication cycle and exhibiting an improved safety profile are of particular interest. The recent marketing of raltegravir (RAL), the first approved HIV integrase inhibitor, seems to fit these requirements. 2,3 RAL prevents the integration of the viral genome into the cellular chromosomes. It has demonstrated high antiviral potency and a good safety profile, both in and outside clinical trials. 4 –8 However, as with many other antiretroviral drugs, if viral suppression is not complete under drug therapy, the virus may select for resistance mutations, which ultimately lead to treatment failure.
RAL resistance mutations are selected within the HIV integrase gene, generally around the catalytic domain (aa 50–212). The pivotal BENCHMRK trials evaluated the efficacy and safety of RAL in HIV-infected patients with prior antiretroviral exposure, and so far constitute the major source of information about RAL resistance. At week 48, 105 (23%) of 462 RAL-treated patients experienced virological failure. 9 Of 94 integrase genotypes obtained, 68% showed RAL-associated resistance mutations at codons 92, 143, 148, and 155, while no changes compared to baseline were seen in 27% of subjects. A few patients selected other integrase changes with unclear phenotypic impact. From weeks 48 to 96, another 5% of patients who remained in the study failed virologically. 10 Again, primary RAL resistance-associated mutations were found in only 64% of them. Altogether, RAL failures were associated with selection of N155H (42%), Q148H/R/K (28%), Y143C/R (10%), and/or E92Q (2%), while 32% of patients did not select for any known RAL resistance-associated mutation.
Given that the clinical experience with integrase inhibitors is still scarce, and the information regarding selection of drug resistance mainly derives from a limited number of patients enrolled in clinical trials, we prospectively undertook the examination of all consecutive virological failures in patients treated with RAL-containing antiretroviral regimens outside clinical trials during the first year of the marketing of the drug in Spain.
Materials and Methods
Study population
All HIV-1-infected individuals treated in Spain during the year 2009 with an RAL-containing antiretroviral regimen at 22 HIV clinics who experienced virological failure (>50 HIV-RNA copies/ml), confirmed in two separate specimens, were identified. Only the first sample with detectable viremia after having achieving undetectability on RAL or specimens collected at months 4–6 of therapy if they never reached undetectable viremia were chosen. A case report form specially designed for this study was filled out and a plasma specimen was shipped to the reference laboratory at Hospital Carlos III, Madrid. Information on viral load, CD4 counts, and antiretroviral drugs before beginning RAL and at the time blood was drawn was recorded. Participants were further asked to send plasma specimens, when possible, collected at baseline and during the previous period when RAL was completely suppressing viremia. A control group of patients who sustained viral suppression on RAL was similarly examined. Most participating centers belonged to RIS (Red de Investigación en SIDA), the government funded Spanish AIDS research network, which involves around 25 HIV clinics across the country and whose main characteristics have already been described elsewhere. 11 The study was approved by the hospital ethics committee.
Drug resistance
The analysis of the HIV integrase gene was performed using an in-house polymerase chain reaction (PCR) protocol, with primers and conditions previously described elsewhere. 12 Four changes were considered as the major RAL resistance-associated mutations: E92Q, Y143H/R/C, Q148H/R/K, and N155H. In addition to the integrase gene, the HIV-1 protease and reverse transcriptase regions were amplified and sequenced using the ViroSeq HIV-1 genotyping system (Celera Diagnostics, Alameda, CA). The genotypic sensitivity score (GSS) of the backbone regimen was calculated using the RIS genotypic interpretation algorithm, 13 which assigns 1, 0.5, or 0 points to distinct mutations, considering single drugs as susceptible, partially resistant, or resistant, respectively. In addition to protease and reverse transcriptase inhibitors, the GSS also considered the use of entry inhibitors such as enfuvirtide and maraviroc.
Pharmacokinetics
RAL trough concentrations (C t) were measured in 100 μl of plasma using a modified validated high-performance liquid chromatography mass spectrometry (HPLC/MS/MS) method, which has previously been described elsewhere. 14 RAL C t<10 ng/ml was considered as undetectable. Plasma specimens from patients failing RAL-containing regimens were obtained at the time of first failure and, when available, during the previous period of undetectable viremia. All blood samples were drawn right before the morning dose of RAL. In addition, RAL C t was measured in a group of RAL-treated patients who sustained viral suppression for at least 1 year. When more than one specimen had been collected during the period of suppressed viremia under RAL therapy, median values were calculated to minimize intrapatient variability.
Statistical analysis
All results are expressed as absolute values and percentages, and median and interquartile ranges (IQR). Comparisons between groups were performed using nonparametric tests, either Mann–Whitney or Chi-square for independent subjects and Wilcoxon signed rank for longitudinal studies. All statistical analyses were performed using SPSS v.15 (SPSS Inc., Chicago, IL).
Results
Patients failing RAL
A total of 106 patients experiencing virological failure under an RAL-containing regimen were identified. The first virological failure was recognized after a median length of RAL therapy of 10.4 (5.7–14.4) months. The median viral load at failure was 3.5 (2.5–4.4) log HIV-RNA copies/ml and the median CD4 count was 295 (145–476) cells/mm3. The HIV-1 subtype was clade B in 90% of patients.
Before beginning RAL, 36.1% of the patients had undetectable plasma viremia. The reasons for switching to RAL were mainly driven by intolerance to previous regimens (i.e., gastrointestinal disturbances, dislipidemia, etc.), as in other recent switch studies. 6,7,15 In the remaining patients with detectable viremia at the time of initiating RAL, the drug was used as salvage therapy in an attempt to regain complete viral suppression, and the median viral load was 4.08 (2.67–4.89) log HIV-RNA copies/ml and the median CD4 count was 324 (159–476) cells/mm3.
Overall, 53.5% of patients who begun RAL included in their regimen two nucleoside analogues, with the most common combinations being tenofovir–emtricitabine (42.6%) and abacavir–lamivudine (10.9%). Darunavir was the most common protease inhibitor given along with RAL, prescribed in 39.6% of patients. Atazanavir was taken by 5% of the rest and 12.9% received other protease inhibitors. Maraviroc was taken by 11.9% of patients and 9.9% received enfuvirtide. The median GSS of the backbone regimen was 2 (1–2). Table 1 records the main baseline characteristics of the study population.
Main Characteristics of Patients Failing Raltegravir
RAL, raltegravir; GSS, genotypic sensitivity score (number of active drugs); C t, plasma trough concentrations.
Baseline integrase polymorphisms
The viral integrase genetic region could be sequenced in 40 baseline specimens collected from patients who subsequently failed RAL. As expected, no primary RAL resistance mutations were identified. However, secondary changes at positions associated with resistance to integrase inhibitors were identified occasionally. In 23 samples other changes were found: eight I203M, seven S230N, two L68I/V, one E157Q, and one Q146K. Each polymorphism appeared alone, except for four patients who harbored I203M+S230N (2) or L68V+I203M (2).
Raltegravir resistance mutations
Integrase sequences could be obtained from 89 (84%) of 106 RAL failures. The viral load in the remaining 17 samples was very low [median of 2.5 log HIV-RNA copies/ml (2.3–3.5)], which most likely precluded successful amplification. Overall, 30 (33.7%) out of the 89 genotyped patients showed primary RAL resistance-associated mutations. They were as follows: 15 N155H, eight Q148H/R, three Y143R/H, and one E92Q. In addition, three patients harbored complex patterns of mutations, including more than one primary change: one N155H+E92Q, one N155NH+Q148QH+G140GS, and one N155H+Y143YH.
Another nine (10.1%) patients selected only secondary RAL resistance changes H51Q, M154I, N155P, T66TR, L74M/I, and G163E/Q. The remaining 50 patients did not select any change at the integrase gene. Table 2 records all changes selected upon RAL failure in the study population.
Distribution of Mutational Patterns in 30 Patients Who Failed Raltegravir with Major Integrase Resistance Mutations
In two patients who failed RAL, a switch to another regimen did not lead to complete viral suppression. One of these subjects selected N155H+V151I at week 34 and added L74M at week 42 of RAL therapy. After discontinuing RAL at week 68 and remaining on the other drugs, a sample taken 16 weeks later showed reversion to wild type at the integrase region. No integrase resistance mutations were similarly noticed in a sample collected 62 weeks later during a viremic episode. The other patient who failed RAL selected Y143R+T97A at week 49 of therapy. He stopped RAL at week 52 but remained on the other drugs and a sample drawn 9 weeks later showed reversion to wild type. However, specimens collected thereafter during viremic periods from weeks 15 to 109 showed a complex evolution alternating wild-type and RAL-resistant genotypes, including amino acid mixtures Y143H/R/C.
Raltegravir failures with and without resistance mutations
A high proportion (66.2%) of patients failing RAL did not select any known primary RAL resistance-associated mutation. When comparing these patients with those who selected primary RAL-resistant genotypes, at failure they had significantly greater viral load [4.04 (3.22–4.69) vs. 3.12 (2.55–3.93) log HIV-RNA copies/ml, p=0.006] and lower CD4 counts [218 (90–354) vs. 441 (172–618) cells/mm3, p=0.008]. Moreover, a significantly greater proportion of patients without major resistance mutations had initiated RAL with detectable viremia compared to the rest (78.4% vs. 45.8%, p=0.013) and included in their regimen a protease inhibitor (71.2% vs. 25.9%, p<0.001). As shown in Table 1, the number of active drugs in the backbone regimen was also greater in patients who failed without major RAL resistance mutations than in the rest [2 (2–3) vs. 1 (0–1), p<0.001].
Raltegravir plasma Ct concentrations
Plasma trough concentrations of RAL could be measured in 66 patients at the time of RAL failure. The median value was 77.4 (0–276.6) ng/ml. In 21 of these patients, RAL C t was also measured during the previous period of undetectable plasma viremia. Then, the median value was 221.8 (75.7–653.7) ng/ml, significantly greater than at the time of failure (p<0.001). In a control group of 37 patients who kept complete viral suppression on RAL, C t was measured in a median of three separate longitudinal specimens per patient collected during a median of 13.4 (9.3–19.5) months; the median RAL C t was 548.7 (215.3–906.7) ng/ml (Fig. 1).
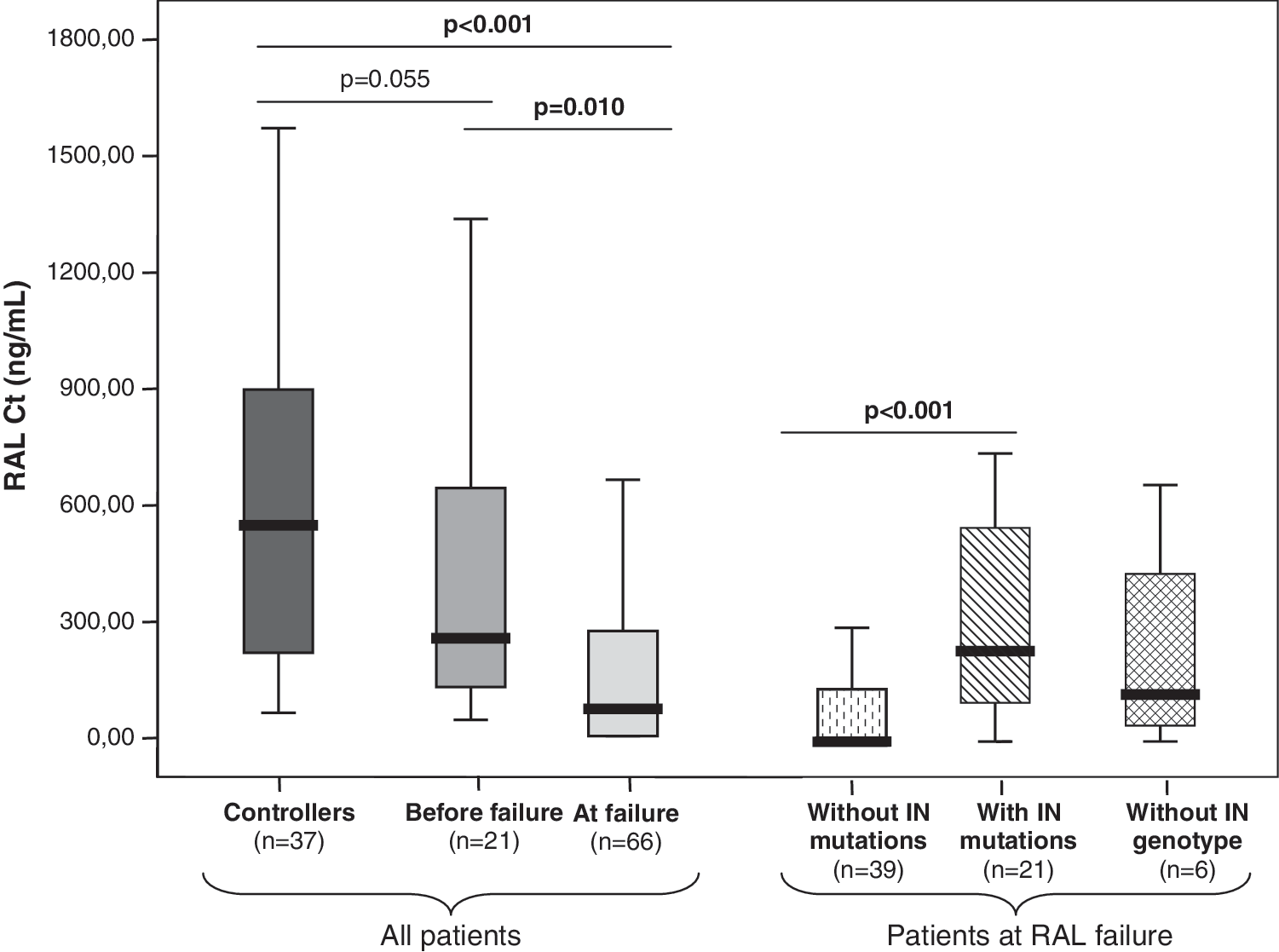
Median raltegravir plasma trough concentrations in the study population.
Upon failure, median RAL C t was lower than in controllers (p<0.001) and on samples collected in the same individuals before failure (p=0.010). There were no significant differences between RAL C t in controllers and in specimens collected before failure in patients who showed virological rebound thereafter, although there was a clear trend toward lower RAL C t in the latest (p=0.055).
Patients who failed without selecting major RAL resistance-associated mutations showed significantly lower median RAL C t than the subset depicting resistance changes [0 (0–135.1) vs. 233.9 (100.9–564.9) ng/ml, p<0.001]. Moreover, in contrast with the latest group, a high proportion of the former patients had undetectable RAL plasma levels (64.1% vs. 0%, p<0.001), while in subjects failing with detectable RAL plasma levels, there were no significant differences in median C t when comparing those with and without RAL resistance mutations [233.9 (100.9–564.9) vs. 221.2 ng/ml (120.5–489.7), p=0.678].
Overall, 25 out of 39 patients (64.1%) without major RAL resistance mutations at failure had undetectable RAL plasma levels, while RAL plasma concentrations were adequate in 14 (35.9%) of them. Moreover, median viral load at failure was greater in patients with undetectable RAL levels than in those with adequate RAL exposure [4.4 (3.8–4.9) vs. 3.5 (2.3–4.3) HIV-RNA log copies/ml, p=0.003]. Only three out of 14 patients failing without major RAL resistance mutations despite harboring adequate drug levels showed secondary changes at the integrase, which were G163E, G163Q, and T66R.
In the longitudinal analysis of 21 patients, RAL C t was measured before and upon virological failure (Table 3). During the period of complete viral suppression, median RAL C t was 221.8 (75.7–653.7) ng/ml and declined to 183.88 (8.5–292.1) ng/ml at the time of failure (Wilcoxon signed ranks test p=0.149). Similar results were obtained when patients were stratified according to selection of RAL resistance mutations. However, half of the patients (5 out of 10) without selecting RAL resistance mutations showed undetectable RAL concentrations upon failure, while median values of 409.1 (91.3–797.1) were recognized during the prior period with suppressed viremia (Table 4). In contrast, RAL plasma exposure had always been detectable in patients who selected RAL resistance changes at failure and median RAL C t did not change significantly [137.1 (34.5–281.5) vs. 208.9 (90.4–299.6) ng/ml, respectively] when comparing both periods (Fig. 2).
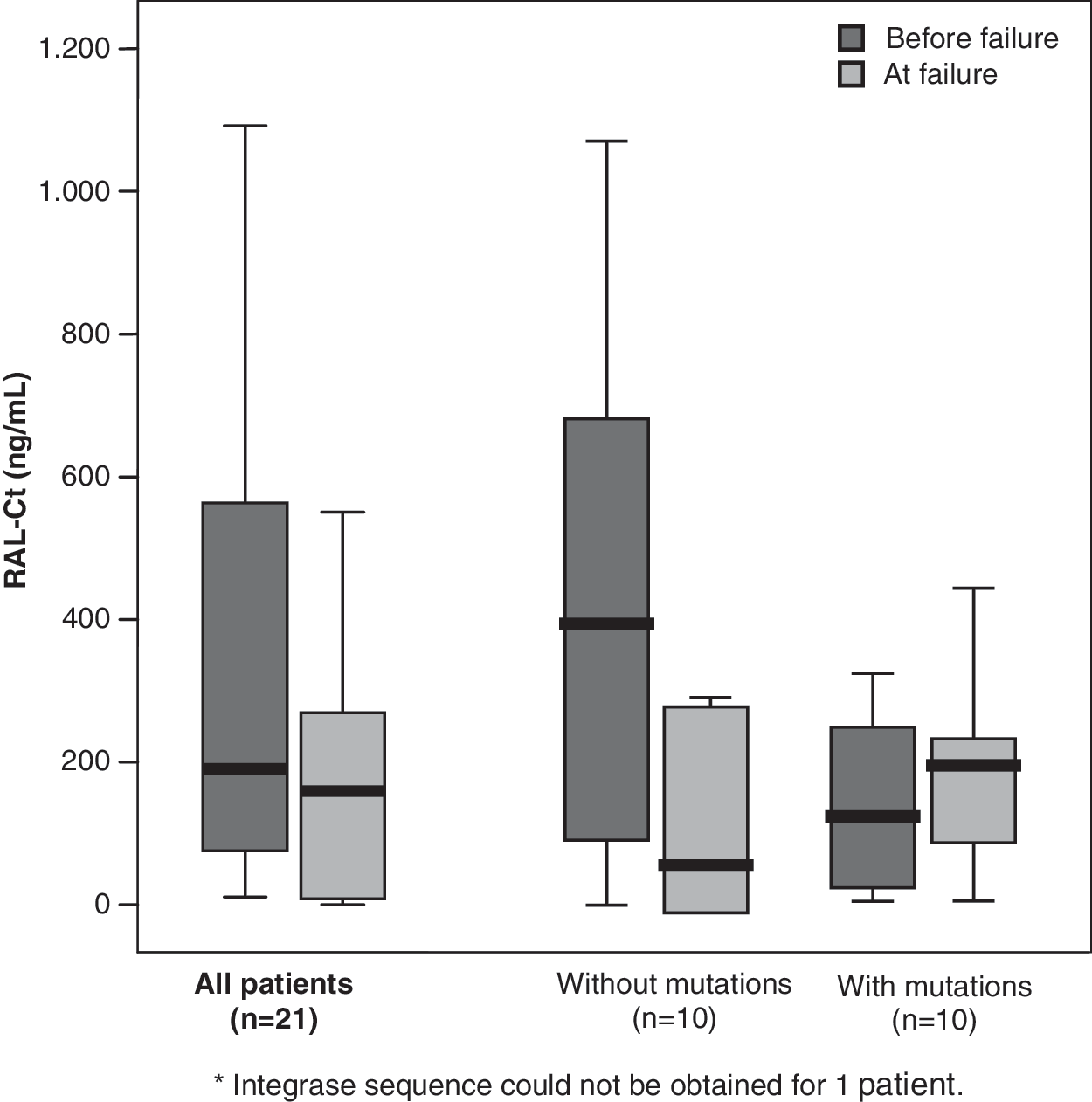
Median raltegravir plasma trough concentrations in the longitudinal study.
Main Characteristics of Patients Who Failed Raltegravir and Had Longitudinal Follow-up
VL, viral load (log10 HIV-RNA copies/ml); CD4, CD4 count (cells/mm3); GSS, genotypic sensitivity score; 3TC, lamivudine; ABC, abacavir; FTC, emtricitabine; TDF, tenofovir; ATV, atazanavir; AZT, zidovudine; SQV, saquinavir; DRV, darunavir; LPV, lopinavir; /r, boosted with ritonavir; NA, not available.
Median Raltegravir Plasma Trough Concentrations (ng/ml) Before and Upon Raltegravir Failure
A readable integrase sequence could not be obtained in one patient.
Discussion
This study reports the rate of RAL resistance mutations in a large series of HIV-infected patients failing the drug in multiple sites in Spain following its approval. To our knowledge this is the largest group of RAL failures examined outside clinical trials so far. A total of 89 viral integrase sequences could be obtained from 106 early virological failures on RAL. Interestingly, up to 66.3% of patients did not harbor viruses with any major RAL resistance-associated mutation, although nine of them depicted secondary changes, as H51Q, M154I, N155P, T66TR, L74M/I, or G163E/Q. The presence of these polymorphisms at the integrase does not seem to directly impact raltegravir susceptibility unless they accompany primary changes. What polymorphisms might influence, however, is a distinct genetic barrier to resistance and/or a different resistance pathway (i.e., codon 155 vs. 148). 9,12 The remaining 30 patients (33.7%) selected RAL resistance mutations including, by order of frequency, changes N155H, Q148H/R, Y143R, and E92Q. In three of them complex mutational patterns including more than one primary RAL resistance mutation were observed.
Our findings are in contrast to prior reports that have found a greater rate of RAL resistance changes upon failure. For instance, the sum of the seven most recent publications on RAL included a total of 58 patients, of whom 46 (79%) selected major RAL resistance changes (25 N155H, 16 Q148H/R, and five Y143R/H/C). 16 –22 The high proportion of our patients failing RAL without resistance mutations can be explained for several reasons. First, ours were early virological failures, and therefore drug exposure under ongoing viral replication was only recent, which might have minimized the time needed for resistance changes to emerge. In a similar way, a high proportion (8/10) of drug-naive patients recruited in the p004 and STARTMRK trials who experienced failure displayed lamivudine or emtricitabine resistance mutation M184V for a while before the emergence of RAL resistance changes. 23
Second, up to 58% of our patients failed RAL along with ritonavir-boosted protease inhibitors and it is well known that the latest reduce selection of drug resistance changes. 13 Third, poor drug adherence seemed to explain most of our cases (25/39; 64.1%) of virological failure on RAL in the absence of drug resistance mutations, given that RAL C t was undetectable in most instances. Reinforcing this hypothesis, most sequences belonging to the viral reverse transcriptase and protease in these patients did not show resistance mutations to other antiretroviral drugs. Moreover, median viral load was greater and CD4 counts lower in these patients compared to those failing RAL with resistance changes, indirectly suggesting that lack of drug pressure rather than escape with drug-resistant strains was occurring.
A subset of our patients failed RAL despite adequate drug exposure in the absence of RAL resistance mutations. In most of them, plasma viremia was relatively low. In this situation, it might be argued that the pharmacokinetics of the drug in specific body compartments could limit drug exposure. If RAL does not reach adequate drug levels in certain body compartments, the virus might escape and replicate there without requiring selection of resistance mutations. In this regard, although RAL levels seem to be adequate in semen 24 and in cervicovaginal fluid, 25 only half of the patients show RAL inhibitory concentrations in the cerebrospinal fluid. 26 An alternative explanation for escaping adequate drug pressure in the absence of resistance mutations might derive from an unexpected reduced RAL susceptibility in the presence of changes in viral genes other than the integrase. This assumption is based on the well-known interactions between the reverse transcriptase and the integrase 27 and the report of an interplay between drug resistance mutations in these regions. 28,29 However, further studies have not confirmed this hypothesis 30,31 and the current belief is that reverse transcriptase or protease resistance-associated mutations do not influence RAL susceptibility.
The measure of total plasma concentrations of RAL as a surrogate of intracellular exposure may be controversial given the mechanism of action of the drug. 32 However, a recent study has demonstrated that both concentrations correlate quite well. 33 The current main limitation is the lack of establishment of a well-defined therapeutic range for RAL and the recognition of substantial interindividual and intraindividual variability. 14,34,35 In our study, significant differences in RAL C t were noticed when comparing patients controlling viremia and those experiencing viral rebound. Moreover, a strong trend toward lower RAL plasma concentrations was noticed when examining RAL levels before and upon failure in samples collected longitudinally from single individuals. In a similar way, Scherrer et al. observed that although RAL levels did not predict virologic failure, up to 60% of failures occurred in patients with very low RAL concentrations. 22 This association, however, has not been confirmed by others. 14,35 Given all these considerations, at this time the main conclusion derived from measuring RAL plasma concentrations in our study is the strong association between viral rebound and undetectable RAL plasma levels, acting as a surrogate of poor drug adherence. Failures were mainly explained by lack of treatment compliance.
Finally, after RAL withdrawal in two patients, RAL resistance mutations disappeared, which indirectly suggests that these changes impair viral fitness. Similar observations have been reported by others. 36,37 Altogether, our findings indicate that early virological failures under RAL-containing regimens may benefit from drug resistance testing. If no integrase mutations are recognized, integrase inhibitors might still be kept as an option for future rescue interventions. On the other hand, if integrase resistance changes are documented, an early switch to other drug families may be warranted to avoid the evolution and accumulation of further integrase resistance changes, which may compromise to a larger extent the activity of second-generation integrase inhibitors. 38
We should acknowledge several limitations of our study, such as the small number of baseline samples analyzed and the absence of baseline samples for the control group. Moreover, we did not determine if any substitution at the integrase that has not been previously related to RAL resistance emerged in the 40 patients who had baseline samples. For a new antiretroviral such as RAL, the absence of resistance mutations in virologic failures might be due to the lack of a complete understanding of genotypic correlates of resistance. In this sense, studies are in progress to characterize those specimens phenotypically.
In summary, a high proportion of patients failing RAL do not select any resistance mutation at the integrase gene. The measurement of RAL plasma concentrations revealed that most of these patients with a lack of resistance mutations were in fact noncompliant. Previously reported major resistance mutations N155H, Q148H/R, Y143R, and E92Q were found in most failing subjects with adequate RAL plasma levels, although some individuals exhibited only secondary integrase resistance changes or no mutations at all. The reasons for these failures remain unclear and deserve further investigation. An interplay between drug adherence, viral fitness cost of resistance mutations, and the extent of viral replication may account for the lack of universal recognition of RAL resistance-associated mutations in patients experiencing early virological failure on RAL-containing regimens.
Spanish Integrase Resistance (SINRES) Group
J. Pedreira (Hospital Juan Canalejo, La Coruña); J.A. Iribarren (Hospital de Donostia, San Sebastián); J. Solá (Hospital de Navarra, Pamplona); M.J. Téllez and V. Estrada (Hospital Clínico San Carlos, Madrid); S. García and J.R. Arribas (Hospital La Paz, Madrid); l. Santos (Hospital de la Princesa, Madrid); P. Miralles (Hospital Gregorio Marañón, Madrid); J.E. Losa (Hospital Universitario Fundación de Alcorcón, Madrid); M. Cervero (Hospital Severo Ochoa, Madrid); J. Sanz (Hospital Príncipe de Asturias, Alcalá); C. Barros (Hospital Universitario, Móstoles); M. Górgolas (Fundación Jiménez Díaz, Madrid); E. Ribera (Hospital Vall d'Hebrón, Barcelona); C. Vidal and M. Riera (Hospital Son Dureta, Palma de Mallorca); C. Gómez (Complejo Hospitalario, Toledo); A. Chocarro (Virgen de la Concha, Zamora); M.J. Galindo (Hospital Clínico Universitario, Valencia); F. Gutiérrez (Hospital General Universitario, Elche); I. Viciana and J. Santos (Hospital Virgen de la Victoria, Málaga); M. Omar (Hospital Ciudad de Jaen, Jaen); A. Collado and C. Gálvez (Hospital Torrecardenas, Almeria); A. Lozano and J.M. Fernández (Hospital Poniente, Almería); V. Gutiérrez-Ravé (Hospital General de Motril, Granada); M.A. López Ruz, C. Hidalgo, and J. Pasquau (Hospital Universitario Virgen de las Nieves, Granada); F. García, V. Guillot, and J. Hernández-Quero (Hospital Universitario San Cecilio, Granada); J.A. Pineda (Hospital de Valme, Sevilla); C. Garrido, L. Anta, C. de Mendoza, and V. Soriano (Hospital Carlos III, Madrid).
Footnotes
Acknowledgments
This work was supported in part by grants from Fundación Investigación y Educación en SIDA (IES), Red de Investigación en SIDA (RIS, ISCIII-RETIC RD06/006), Fondo de Investigación Sanitaria (FIS project PI06/1826 and CP06/0284), Agencia Laín Entralgo, and NEAT
Author Disclosure Statement
No competing financial interests exist.