
Abstract
Recombination between HIV-1 subtypes B and F has generated several circulating and unique recombinant forms, particularly in Latin American areas. In Italy, subtype B is highly prevalent while subtype F is the most common pure non-B subtype. To investigate the recombination pattern in Italian BF recombinant viruses, we characterized full-length sequences derived from 15 adult patients, mostly Italian and infected by the heterosexual route. One of the BF mosaics was a CRF29, three sequences clustered with low bootstrap values with CRF39, CRF40, and CRF42. With the exception of the CRF29-like sequence, the other recombination patterns were unique, but two possible clusters were identified. Analysis of the gp120 V3 domain suggested a possible link with subtype F from Eastern Europe rather than from Latin America, favoring the hypothesis of local recombination between clade B and F viruses over that of import of BF recombinants from Latin America. HIV-1 subtypes B and F appear prone to generation of unique recombinants in Italy, warranting epidemiological surveillance and investigation of a possible clinical significance.
Human immunodeficiency virus type 1 (HIV-1) is characterized by an extremely broad genetic diversity, caused by high mutation and recombination rates. Currently, HIV-1 group M (“Major”), which is responsible of the worldwide pandemic, is divided into pure subtypes A–D, F–H, J, and K (with subtypes A and F further subdivided into subsubtypes A1–A4 and F1–F2, respectively) as well as 48 circulating recombinant forms (CRFs) of varying epidemiological significance, and untold numbers of unique recombinants that are not known to have spread to many patients. Subtype B has long been predominant in Western European countries but the prevalence of non-B strains has been increasing significantly in past years as a consequence of recent migration waves from Africa, Eastern Europe, and South America. 1 Continuous surveillance of HIV-1 molecular epidemiology plays a critical role in the understanding of genetic diversity of HIV-1 and for research purposes such as vaccine development. In clinical practice, pol-based subtype assignment is usually accomplished as a by-product of genotypic antiretroviral resistance testing.
A homebrew HIV-1 genotyping assay has been established and offered as a public health service at the HIV Monitoring Laboratory (HML), Department of Molecular Biology, University of Siena, Italy since 1995. 2 The laboratory has been serving a number of clinics for analysis of drug resistance mutations accumulating around 10,000 protease and reverse transcriptase sequences from more than 4000 patients. In addition, some hundred partial gag and env sequences have been obtained for research studies. A recent survey on the whole HIV-1 sequence database at the HML revealed 85% of subtype B sequences and 15% of non-B subtypes, mainly CRF02_AG, F1, C, and A1, based on the pol region. 3 In some cases, assignments of subtype based on the pol and env regions were not in agreement. 4 These discrepancies mostly involved subtypes B and F1. In addition, other sequences showed BF1 mosaics within the individual pol or env region. Since BF recombinants have been previously reported in Latin America, 5 –11 we were interested in analyzing BF mosaic forms circulating in Italy.
Samples from 15 adult patients (12 males, three females) collected in four hospitals in the Tuscany and Umbria regions were selected for near full-length sequencing based on peripheral blood mononuclear cell (PBMC) DNA availability and identification of a BF pol sequence or discordant BF pol and env. Twelve patients were Italian, two were Brazilian patients living in Italy, and 1 was a Tunisian patient; most of the patients acquired HIV-1 infection via sexual intercourses; only one patient contracted HIV-1 infection through drug injection (Table 1).
Epidemiological Data of the Patients Included in This Study
Cluster 1 sequences and bcluster 2 sequences are based on phylogenetic tree as shown in Fig. 1.
To obtain full-length sequence information, PBMC DNA was subjected to a 9010-base pair preamplification step 12 followed by multiple nested polymerase chain reactions (PCRs) generating overlapping subgenomic fragments. The LTR region, not comprised in the large outer amplicon, was obtained separately using outer primers MZ28 (coordinates 60–79 in HIV-1 HXB2) and LR56 (1488–1507) and inner primers P221 (87–100) and P82 (1478–1505). PCR products were sequenced on both strands using multiple infrared-labeled primers and sequencing products were resolved with a Licor IR2 dual-laser automated sequencer. PCR and sequencing primer sequences are available on request. Nucleotide sequences were edited and assembled by the DNAstar SeqMan II module.
For phylogenetic analysis, full-length sequences were compared with full-length subtype reference sequence alignments obtained from the Los Alamos National Laboratory HIV Sequence Database (
Phylogenetic analysis was performed with PAUP4b10 software. Neighbor joining and maximum likelihood trees were constructed based on the fittest nucleotide substitution models determined by Modeltest v3.7. The reliability of the tree topology was assessed by bootstrapping with 1000 replicates. The recombination patterns were determined by bootscanning analysis with SimPlot v3.5.1 (
Phylogenetic analysis of the near full-length sequences is shown in Fig. 1. Sample 59211 was identified as a CRF29, while samples 83166, 30638, and 85037 clustered with low bootstrap values with CRF39, CRF40, and CRF42, respectively. Bootscanning analysis showed the high genetic diversity between reference sequences and our BF mosaics, which are mostly characterized by unique recombination patterns. Two distinct clusters of BF sequences shared a similar recombination pattern, as shown in Fig. 2. Each BF cluster may have originated from a common ancestor circulating among people living in the same geographic area. Further recombination events may then have introduced different breakpoints contributing to diversification within the same cluster. This hypothesis is supported by greater sequence similarity among members of cluster 1 and among members of cluster 2 than between clusters or the unique recombinants. Cluster 2 showed a high degree of relatedness and may represent a new circulating recombinant form if complete genomes of other isolates are completed in the future.
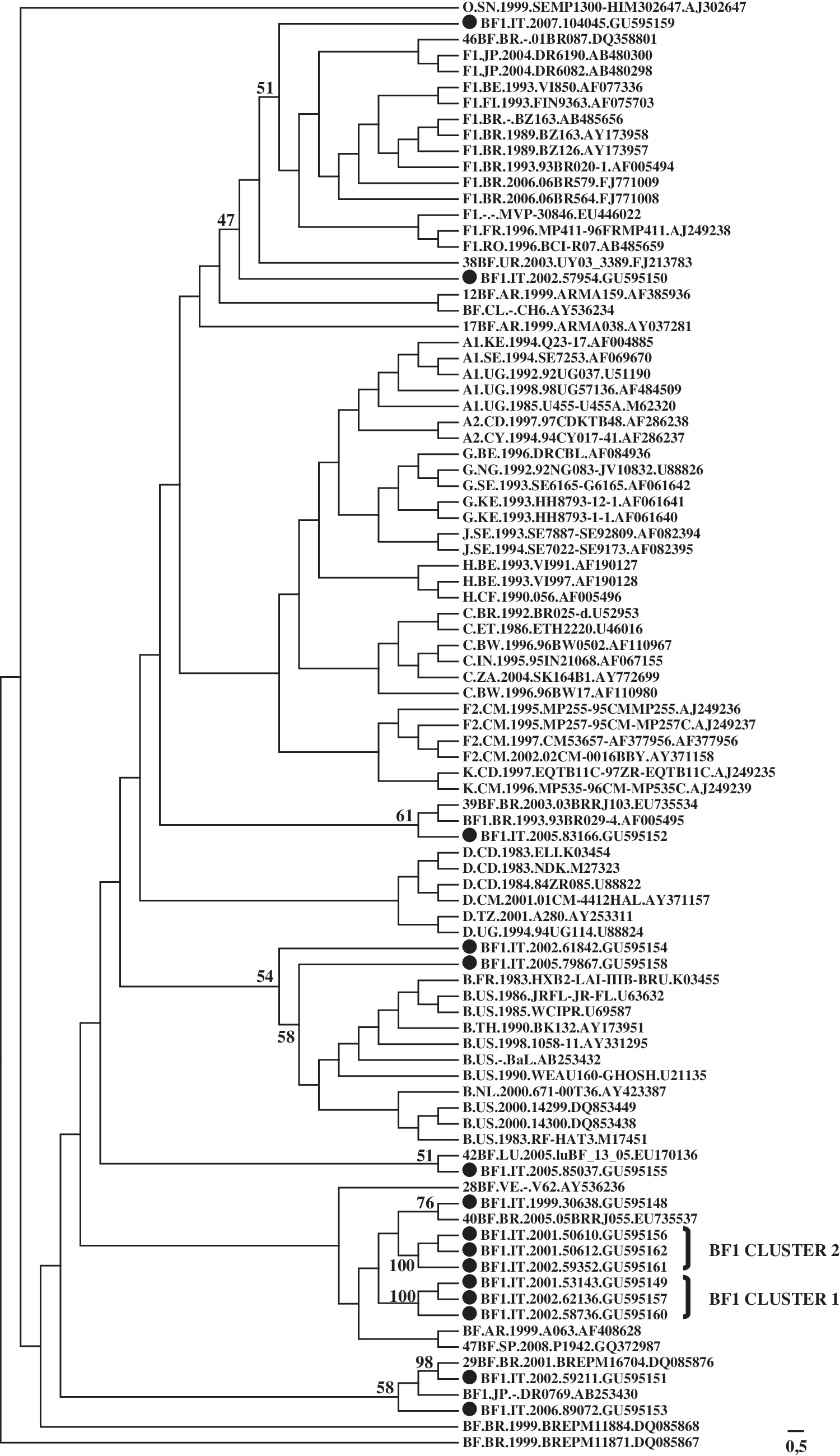
Neighbor-joining tree of near full-length BF recombinant sequences generated with the GTR + I + G maximum likelihood model. Black circles indicate BF recombinants sequenced in this study. Bootstrap values based on 100 replicates are shown at key nodes.
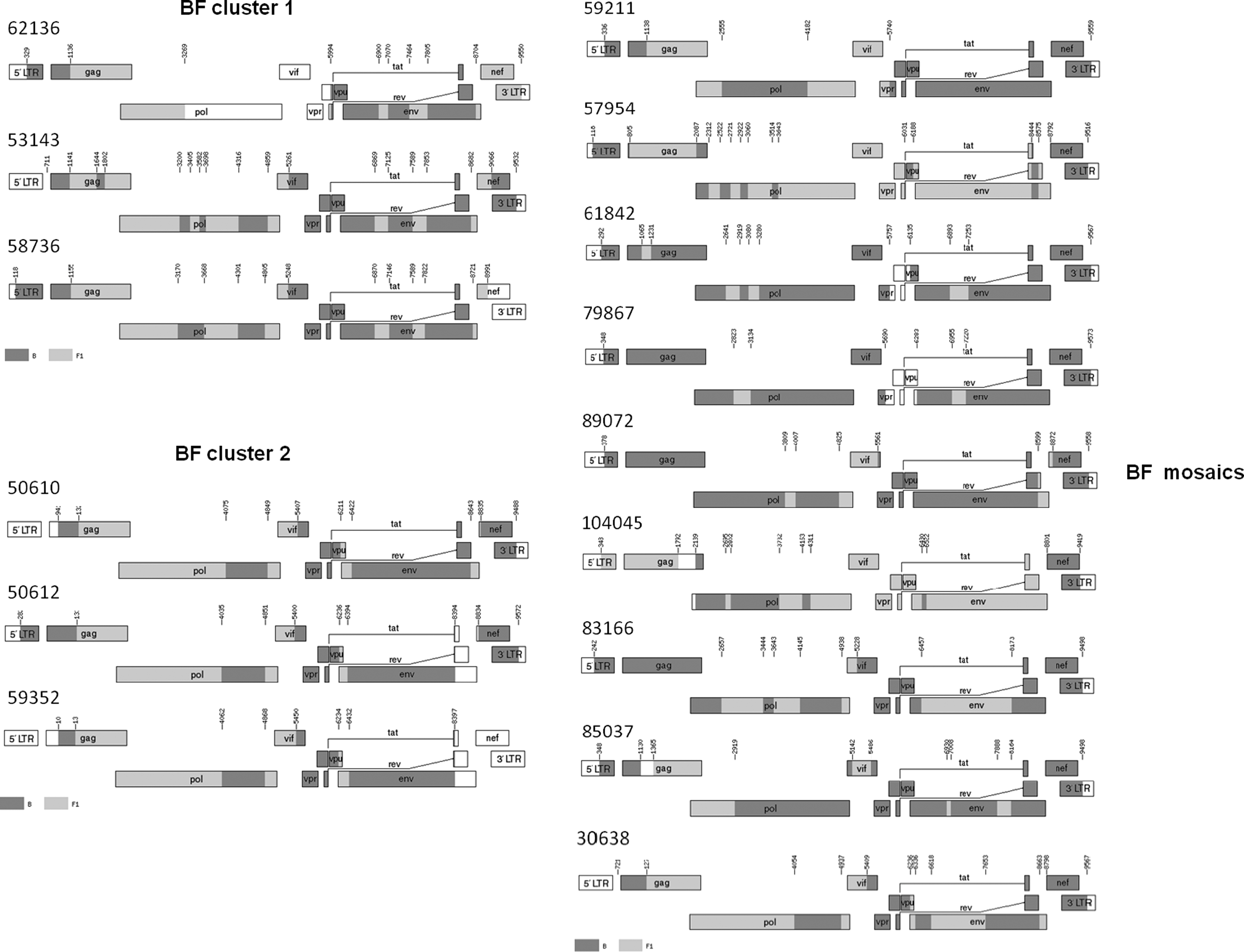
Schematic representation of near-full length genomes of 15 BF recombinant viruses from Italy. Subtype B and F1 regions are shown in dark and light gray, respectively. Genomic regions that were not sequenced are shown in white. Recombination breakpoint positions are referred to the HXB2 nucleotide coordinates. Pictures were generated using the “Recombinant drawing tool” available at the Los Alamos HIV Sequence database website.
BF recombinants where first isolated and characterized in Argentina, where they have been circulating since the mid-1980s,
5
and later in Brazil.
6
The presence of these recombinants and related unique mosaic strains increased over the years, favored by the high number of injecting drug users, sexual contacts, and the cocirculation of subtype B, F, and BF viruses in the same populations.
7,8
Afterward, BF mosaics were described in other Southern America countries
9,10
as well as in European countries such as Spain
13
and Luxembourg (where CRF42_BF has been isolated). Recently, a full-length sequence of a BF recombinant strain with a unique mosaic pattern has been characterized in Italy
14
in a patient with multiple risk factors and found to be phylogenetically related to two other sequences from Brazil. Most of the BF genomes described in this study were acquired via heterosexual contacts. BF mosaics could have been imported into Italy from Southern America or locally generated by independent recombination events between subtype B, which is predominant in Italy, and subtype F1, which is the most frequent non-B pure subtype circulating in Italy. The migration flow from South America to Italy and sexual tourism in Brazil could support the first hypothesis. Although most of the sequences reported here do not seem to be related to the CRFs originated in Brazil or Argentina and other BF sequences found in GenBank (data not shown), phylogenetic analysis of the pol region indicated that the seven BF recombinants with an F1 pol are more similar to the F1 subtype circulating in South America than to that circulating in Eastern Europe (Supplementary Fig. S1; Supplementary Data are available online at
Analysis of the gp120 V3 loop sequence predicted 11 CCR5-tropic and 4 CXCR4-tropic viruses. The V3 crown tetrapeptide was GPGR in 11/15 (73.3%) sequences, GPGQ in 2/15 (13.3%), and GPGG and GPGK in the other two cases. Among nine clade B V3 sequences, the GPGR motif was observed in seven cases, while the other two sequences had GPGG and GPGK motifs. Among six clade F1 V3 sequences, GPGR was found in four cases and GPGQ in the other two samples. None of the samples with subtype B V3 region had the GWGR motif, which is common in clade B viruses circulating in Brazil but not in other countries. 15,16 Interestingly, two of the subtype F1 V3 sequences had the GPGQ crown variant, which is largely prevalent in Romania and is not present in Brazilian F1 (mostly GPGR). Thus, this last finding could support an hypothesis of the local origin of BF recombinants due to the cocirculation of subtypes B and F1.
The appearance of these mosaic structures in Italy can be compared to what occurred in Argentina, where subtype B was predominant before the introduction of subtype F1. Actually, the prevalence of BF recombinants has been increasing in Latin American countries, becoming the most common genetic form and causing the disappearance of the pure subtype F in some areas. 5,15 The high propensity for subtype B and F to recombine could be explained by a higher fitness compared with pure subtype B and F1. 11 To test this hypothesis, extensive in vitro analysis of chimeric viruses is required to investigate the interaction among subgenomic regions from each subtype. 17 In general, the prevalence of BF, as well as other mosaic structures is probably underestimated due to large-scale partial genotyping of clinical samples for antiretroviral resistance testing. Along with the upcoming availability of effective technologies for large-scale sequencing, information on full-length HIV genomes is expected to expand providing an accurate picture of virus diversification in different areas over time.
Sequence Data
GenBank accession numbers of the sequences are GU595148–GU595162.
Footnotes
Acknowledgments
This work was supported by grants from the Italian Ministry of Health (PRIN Grant 200887SYZ5 and AIDS Program Grant 40h81) and from the European Community under the Seventh Framework Program (CHAIN project Grant 223131).
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.