
Abstract
TMC278-C204 (NCT00110305), a 96-week trial of the nonnucleoside reverse transcription inhibitor (NNRTI) rilpivirine (RPV, TMC278) in 368 HIV-1-infected, treatment-naive patients, was extended to investigate long-term safety and efficacy. Week 192 analysis results are presented. This was a long-term follow-up of a Phase IIb, randomized trial. No significant RPV dose-response relationships with respect to the primary endpoint (composite ITT-TLOVR algorithm) were observed at week 48 or 96. All RPV-treated patients were switched to open-label 75 mg qd at week 96 and then to 25 mg qd, the Phase III dose, at approximately week 144 as it gave the best benefit–risk balance. All control patients continued receiving open-label efavirenz (EFV) 600 mg qd. At week 192, 59% of RPV- and 61% of EFV-treated patients maintained confirmed viral load <50 copies/ml (ITT-TLOVR algorithm). The mean changes from baseline in CD4 cell count were similar in both groups (RPV: 210 cells/mm3 vs. EFV: 225 cells/mm3). No new safety concerns were noted between week 48 and 192. In the week 192 analysis, RPV compared with EFV was associated with a lower overall incidence of grade 2–4 adverse events (AEs) at least possibly related to treatment, including rash (p<0.001) and neurologic AEs (p<0.05 Fisher's exact test, post hoc analyses) Incidences of serious AEs, grade 3 or 4 AEs, and discontinuations due to AEs were similar across groups. Increases in total cholesterol, LDL-cholesterol, HDL-cholesterol, and triglycerides were significantly lower with RPV than with EFV. RPV continued to show sustained efficacy similar to EFV at week 192 with a generally more favorable safety profile.
Introduction
T
The next-generation NNRTIs, rilpivirine (RPV, TMC278, EDURANT®) and etravirine (ETR, TMC125, INTELENCE®), showed potent activity against HIV-1 in vitro. 5 ETR has been approved for use in combination with other ARVs in HIV-1-infected treatment-experienced adult patients and, except for rash, has a safety profile comparable to placebo. 6 RPV has been recently approved in the United States and Canada in combination with other ARVs in HIV-1-infected treatment-naive adult patients. 7 The 25 mg dose was investigated in Phase III trials, 8,9 and a once-daily, single-tablet regimen with tenofovir disoproxil fumarate (TDF) plus emtricitabine (FTC) has also been approved in the United States. 10
In the dose-ranging part of this randomized, Phase IIb trial, TMC278-C204 (NCT00110305), with a 96-week treatment period, RPV 25 mg, 75 mg, or 150 mg qd were administered in combination with a dual nucleoside/tide reverse transcriptase inhibitor [N(t)RTI] background regimen in HIV-1-infected treatment-naive patients. 11 All once-daily RPV doses showed potent antiviral efficacy similar to that of the open-label control, EFV 600 mg qd, also given with this background regimen, over 96 weeks. Neurologic and psychiatric AEs and rash were less frequent and serum lipid increases were lower with RPV than with EFV. No significant RPV dose-response relationships for the primary endpoint (composite intent-to-treat-time-to-loss of virologic response [ITT-TLOVR] algorithm) were observed at week 48 or 96. The trial was extended to investigate long-term safety, tolerability, and efficacy of RPV versus EFV. This analysis includes all patients treated for at least 192 weeks, or who discontinued earlier.
Materials and Methods
Patients
The trial was conducted in 54 centers in 14 countries, divided into three regions (Region 1: Asia and South Africa; Region 2: Europe, United States, and Russia; Region 3: Latin America), as previously reported in detail. 11
Eligible patients were aged ≥18 years, had never previously been treated with therapeutic HIV-1 vaccine or received ≤2 weeks of treatment with an N[t]RTI and/or protease inhibitor (PI), had genotypic sensitivity to the selected N[t]RTIs, and had a plasma viral load >5000 copies/ml. The main exclusion criteria included any currently active AIDS-defining illness and prior use of NNRTIs or documented genotypic evidence of NNRTI resistance. 11
Written informed consent was obtained from all patients prior to trial start. The trial protocol was reviewed and approved by the local regulatory authorities and the relevant Independent Ethics Committee/Institutional Review Board. The trial was conducted in accordance with the Declaration of Helsinki.
Trial design and treatments
TMC278-C204 began as a Phase IIb, randomized trial of 368 patients, designed to evaluate the dose-response relationship for efficacy, tolerability, and safety over 96 weeks of three blinded, once-daily RPV doses (25 mg, 75 mg, and 150 mg) administered immediately after breakfast (Fig. 1). 11 Open-label EFV 600 mg qd given in the evening was included as a control. All patients also received a background N[t]RTI regimen of zidovudine/lamivudine (AZT/3TC) or TDF/FTC selected by the site investigators and administered as fixed-dose combinations where available. Randomization was stratified by region and N[t]RTI background. The study was extended to 5 years to collect long-term safety and efficacy data for RPV, and compare with data for EFV.

Trial design: From week 96 to approximately week 144 (range: 131–157 weeks), all rilpivirine (RPV)-treated patients received open-label RPV 75 mg qd and were then switched to RPV 25 mg qd. All EFV-treated patients received open-label EFV throughout the trial. EFV, efavirenz; N(t)RTI, nucleoside/tide reverse transcriptase inhibitor.
Other NNRTIs, PIs, fusion inhibitors, cytochrome P450 3A4 substrates/inducers/inhibitors, and immunomodulators were not allowed during the treatment period. N[t]RTI within-class substitutions and N[t]RTI dose adjustments were allowed only for tolerability reasons.
Trial endpoints
The primary objective of the trial was to evaluate the RPV dose-response relationship for efficacy at week 48. Secondary objectives included, among others, evaluation of safety and tolerability over time. 11
The primary efficacy parameter was the proportion of patients with confirmed viral load <50 copies/ml at week 48 (responders) according to the ITT-TLOVR algorithm. 12 According to this algorithm, patients were considered nonresponders if they had confirmed virologic failure or discontinued prematurely for any reason. Virologic failure for the efficacy endpoint (VFeff) was defined as patients with loss of response, patients who never achieved confirmed viral load <50 copies/ml, or patients who discontinued due to lack of efficacy. Virologic failure for the resistance analysis (VFres) was determined in the ITT population with all available data, regardless of time of failure and reason for discontinuation as long as a patient's virologic profile showed signs of virologic failure, i.e., defined as at least two consecutive viral load values (or last available viral load, in case of early withdrawal) >0.5 log10 copies/ml above nadir (with at least one viral load >500 copies/ml). Secondary parameters included the proportion of patients with viral load <50 copies/ml over time (ITT-TLOVR algorithm) and changes in CD4 cell count from baseline. 11
Assessments
After week 24, patients were seen every 8 weeks. The Division of AIDS criteria were used for grading severity of AEs. 13 Urine and blood samples were collected for urinalysis, biochemistry, hematology, coagulation, immunology (changes in CD4 cell counts), endocrinology, pharmacokinetics, resistance testing, and plasma viral load determinations. The plasma viral load was determined and viral phenotypic and genotypic resistance testing was performed as described previously. 11 The safety and efficacy data were assessed at regular time points by an independent Data and Safety Monitoring Board.
Trial extension
At weeks 48 and 96, no significant RPV dose-response relationships with respect to the primary endpoint (composite ITT-TLOVR algorithm) were observed. RPV 75 mg qd was initially selected for further development in Phase III.
All patients receiving RPV, who in the opinion of the investigator still benefited from their treatment, were switched to open-label RPV 75 mg qd plus investigator-selected N[t]RTIs at week 96 (Extension 1) (Fig. 1). All patients receiving EFV 600 mg qd plus investigator-selected N[t]RTIs continued to receive EFV treatment. In an initial thorough QT trial in HIV-negative volunteers, RPV 75 mg and 300 mg qd doses prolonged the corrected QT (QTc) interval in a dose- and plasma-concentration-dependent manner. In a subsequent QT trial, RPV 25 mg qd had no effect on QTc interval. 14 In TMC278-C204, in the week 96 analysis the incidence of discontinuations due to AEs and rashes and increases in QTcF interval (QT interval corrected according to Fridericia's formula) prolongation were lowest with RPV 25 mg qd. Given the results of the thorough QT study and the lack of significant dose-response relationships for efficacy across dosing groups through week 96, RPV 25 mg qd showed the best benefit–risk balance overall and was selected as the phase III dose. 11 Therefore, at approximately week 144 (range: 131–157 weeks based on the availability of the 25 mg tablet at the study site), all RPV-treated patients were switched (Extension 2) to open-label RPV 25 mg qd, the selected Phase III dose, for the remainder of the total treatment duration of 240 weeks.
Patients not continuing into Extension 1 or Extension 2 had all discontinued and thus were considered nonresponders according to the ITT-TLOVR algorithm at week 192 (even if they had undetectable viral load at the time of discontinuation).
Data analysis and statistics
The sample size was calculated for the week 48 primary analysis (i.e., dose-finding portion of the study). 11 All analyses were performed on the ITT population, regardless of compliance with the protocol.
Fisher's exact test was used to compare the incidence of AEs in the RPV group (all patients receiving RPV regardless of initial dose) versus the EFV group. Treatment comparisons regarding changes from baseline in laboratory values were performed applying a nonparametric Wilcoxon rank-sum test. Comparisons on safety endpoints were post hoc analyses, each performed at 5% significance without adjustment for multiplicity.
Results
Patient disposition, demographic parameters, and baseline characteristics
Of the 515 patients who were screened, 373 were randomized and 368 treated (ITT population). Of these 368 patients, 273 entered Extension 1 (weeks 96–144) and 252 patients entered Extension 2 (weeks 144–240).
At the time of the week 192 analysis, 131/368 (36%) of patients had discontinued the trial (11% increase compared with the week 96 analysis), 11 with no differences between the RPV group and EFV group in the proportion of discontinuations. The most common reasons for discontinuation were AE/HIV-related event (14%; +3% compared with the week 96 analysis), loss to follow-up (5%; +1%), virologic failure (5%; +1%), withdrawal of consent (3%; +0.3%), noncompliance (3%; +1%), completion of week 96, but noncontinuation into Extension 1 (3%; +3%), ineligible to continue (1%; no change), and completion of Extension 1, but noncontinuation into Extension 2 (0.5%; +0.5%). The amendment for Extension 1 was not implemented in Uganda, so 10 patients (eight receiving RPV and two receiving EFV) from this country completed the study at week 96, but did not continue into Extension 1.
Demographic parameters and baseline disease characteristics were generally well balanced across groups and treatments and full details have been reported previously (Table 1). 11 Overall, 75% of patients received AZT/3TC as their background regimen and 25% TDF/FTC. The median (range) duration of treatment in this analysis was 196 weeks (0.1–215 weeks) in the RPV group and 196 weeks (1–212 weeks) in the EFV 600 mg qd group. The median (range) time on the 75 mg qd dose was 48 weeks (2–61 weeks) in Extension 1 and on the 25 mg qd dose was 53 weeks (5–67 weeks) in Extension 2.
Amendment for Extension 1 not implemented in Uganda, so all Ugandan patients were discontinuations and thus counted as failures.
Number of patients switched to RPV 75 mg qd at week 96.
Number of patients switched to RPV 25 mg qd as soon as available on site (approximately week 144; range: 131–157 weeks).
N'=88 for CD4 cell count data.
Hepatitis C infection status was confirmed by hepatitis C virus (HCV) antibody and also qualitative HCV RNA if the test for HCV antibodies was positive or if patients were immunocompromised (CD4 cell count<100 cells/mm3).
RPV, rilpivirine; EFV, efavirenz; ITT, intent-to-treat; N or N', number of patients per treatment group; n, number of patients with individual parameter; HIV-1, human immunodeficiency virus—type 1; CDC, Center for Disease Control and Prevention; RNA, ribonucleic acid; PCR, polymerase chain reaction; FC, baseline fold-change in 50% effective concentration (EC50).
Treatment response
RPV continued to show similar antiviral efficacy to that of EFV over 192 weeks (Table 2 and Fig. 2a). The percentage of patients with confirmed viral load <50 copies/ml (ITT-TLOVR algorithm) at week 192 was 59% in the RPV group and 61% in the EFV group.
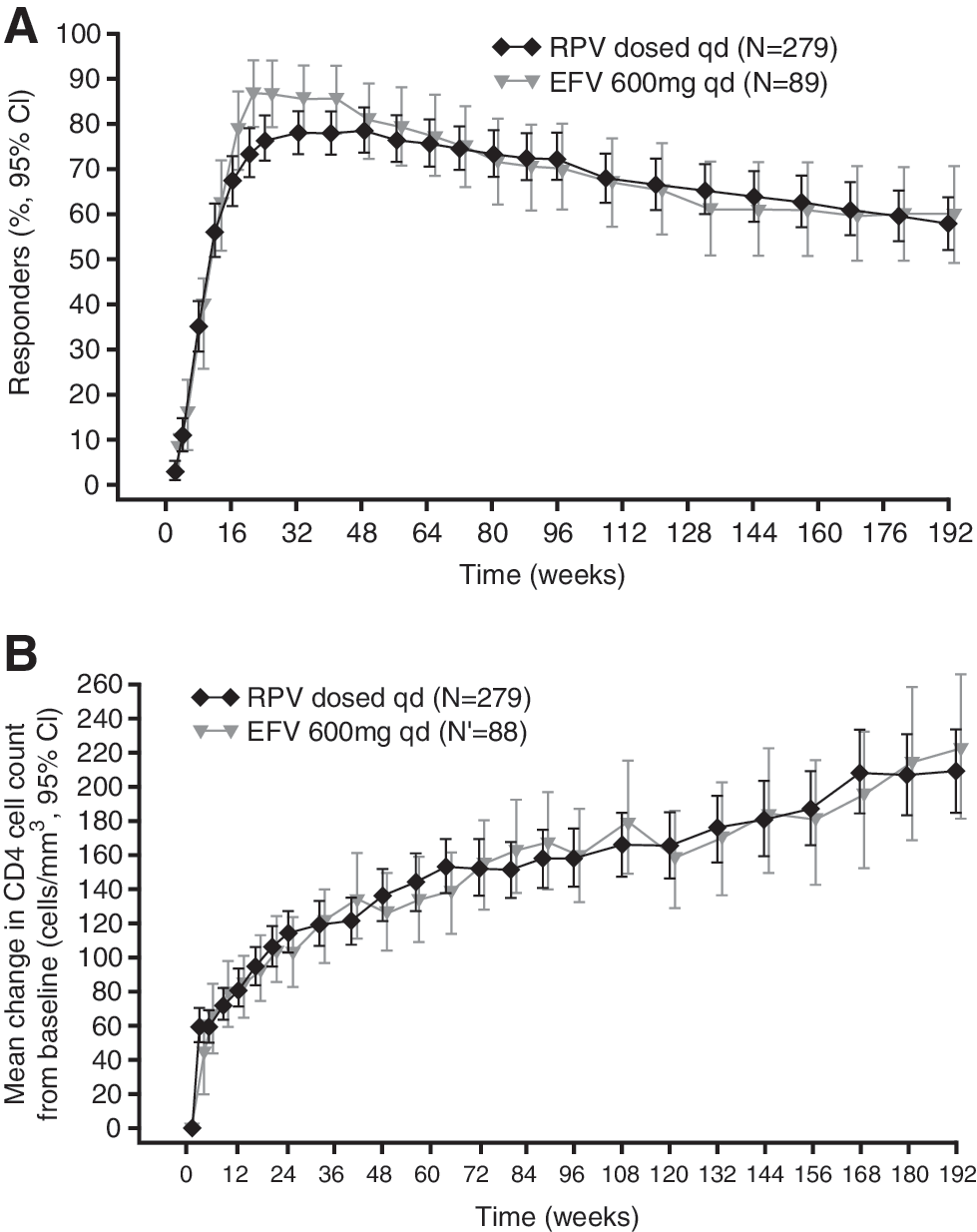
Antiviral efficacy:
N'=88 for CD4 cell count data.
All 95% CIs are derived using normal approximation.
According to time-to-loss of virologic response algorithm.
VFeff is defined as patients with loss of virologic response, patients who never achieved confirmed viral load <50 copies/ml, or patients who discontinued due to lack or loss of efficacy.
Lost to follow-up, noncompliance, withdrew consent, ineligible to continue, sponsor's decision.
Imputed with baseline value after premature discontinuation (i.e., change=0, noncompleter=failure); intermediate missing values imputed using last observation carried forward.
Six RPV patients and one EFV patient switched status from VFeff to another category between week 96 and 144.
One RPV patient and two EFV patients switched status from VFeff to another category between week 144 and 192.
No statistically significant difference between treatment groups.
RPV, rilpivirine, EFV, efavirenz; N or N', number of patients per treatment group; n, number of patients with individual parameter; SE, standard error; CI, confidence interval.
Virologic failures (VFeff and VFres)
VFeff at week 192 was 12% (33/279 patients) in the RPV group versus 8% (7/89 patients) in the EFV group (p=0.3 RPV versus EFV, Fisher's exact test, post hoc analysis). Of the 33 patients in the RPV group who had VFeff at week 192, 20 also met the VFres definition, but the other 13 patients met only the VFeff definition (these patients had two consecutive values >50 copies/ml but did not have an increase of 0.5 log above nadir). Of the 13 patients meeting only the VFeff definition, none discontinued; eight patients had undetectable viral load and five patients had viral load <200 copies/ml at week 192. Of the seven patients in the EFV group in the VFeff analysis, four patients also met the VFres definition with the other three patients meeting only the VFeff definition (these three patients had two consecutives values >50 copies/ml but did not have an increase of 0.5 log above nadir).
Of the RPV patients with treatment response at week 96, 7/204 (3%) up to week 144 and subsequently 10/204 (5%) up to week 192 had VFeff and six patients and one patient, respectively, switched status from VFeff to another category. Therefore, there was a net increase of 10 RPV patients with VFeff at week 192 compared with week 96 (Table 2). Of the EFV patients with treatment response at week 96, 3/63 (5%) up to week 144 and none up to week 192 experienced VFeff with EFV. There were one and two EFV patients, respectively, who switched status from VFeff to another category, so there was no net increase in the number of EFV VFeff at week 192 compared with week 96 (Table 2).
In the week 192 analysis, 11% (31/279) of patients receiving RPV versus 9% (8/89) receiving EFV met the definition of VFres (p=0.7 RPV versus EFV, Fisher's exact test, post hoc analysis). Of the 31 patients in the RPV group, 20 patients met both definitions of virologic failure (VFeff and VFres), with the other 11 patients discontinuing for reasons other than VFeff. Of the eight patients in the EFV group, four met both definitions of VF (VFeff and VFres) and the other four patients discontinued for other reasons than VFeff.
VFres occurred in 17 RPV and 6 EFV patients up to week 96. 11 Of the patients with treatment response at week 96, 3/204 (1%) up to week 144 and subsequently 11/204 (5%) up to week 192 were VFres for RPV, while 2/63 (3%) and none, respectively, experienced VFres with EFV.
Resistance analysis
In the RPV group, genotypic data were available for 30/31 patients with VFres. The proportion of patients with VFres who developed at least one treatment-emergent reverse transcriptase (RT) resistance-associated mutation (RAM) was 70% (21/30) in the RPV group and 88% (7/8) in the EFV group. The most frequent emerging RT RAMs in the RPV group were the NNRTI RAMs E138K and K101E, and the N[t]RTI RAMs M184V and M184I, occurring in 23% (7/30), 20% (6/30), 33% (10/30), and 10% (3/30) of the patients with VFres in the RPV group, respectively. The NNRTI RAMs K103N (3/8, 37.5%) and V106M (1/8, 12.5%) were seen in patients with VFres in the EFV group. No N[t]RTI RAMs, including M184I/V, were observed in the EFV group. The proportion of patients with more than one NNRTI and/or N(t)RTI RAM was 43% (13/30) in the RPV group and 0% in the EFV group.
Immunologic response
The mean and median changes from baseline in CD4 cell count continued to increase over the duration of the trial from week 96 to 192. Mean increases in CD4 cell count were similar in the RPV (210 cells/mm3) and EFV groups (225 cells/mm3) (Table 2 and Fig. 2b).
Safety results
Safety analyses were performed using all available data, including beyond week 192. The week 192 analysis showed that RPV was well tolerated over the extended treatment period. The incidence of any grade 2–4 AE at least possibly related to NNRTI treatment was lower with RPV than with EFV (24% versus 44%, respectively; p<0.01 Fisher's exact test, post hoc analysis). The most commonly reported individual grade 2–4 AEs occurring in ≥2% of patients in either group (excluding laboratory abnormalities reported as an AE) were nausea, dizziness, headache, abnormal dreams/nightmare (grouped term), depression, dyspepsia, asthenia, somnolence, vertigo, and any rash (grouped term) (Table 3). There were no new safety concerns, with no new types of AEs or noteworthy increases in incidence of AEs at least possibly related to RPV or EFV between weeks 48 and 192. Figure 3 shows the incidence of any AEs at least possibly related to investigational treatment (all grades; Fig. 3a and grades 2–4; Fig. 3b), between weeks 48 and 192, showing the highest incidence was in the first 48 weeks in both groups.
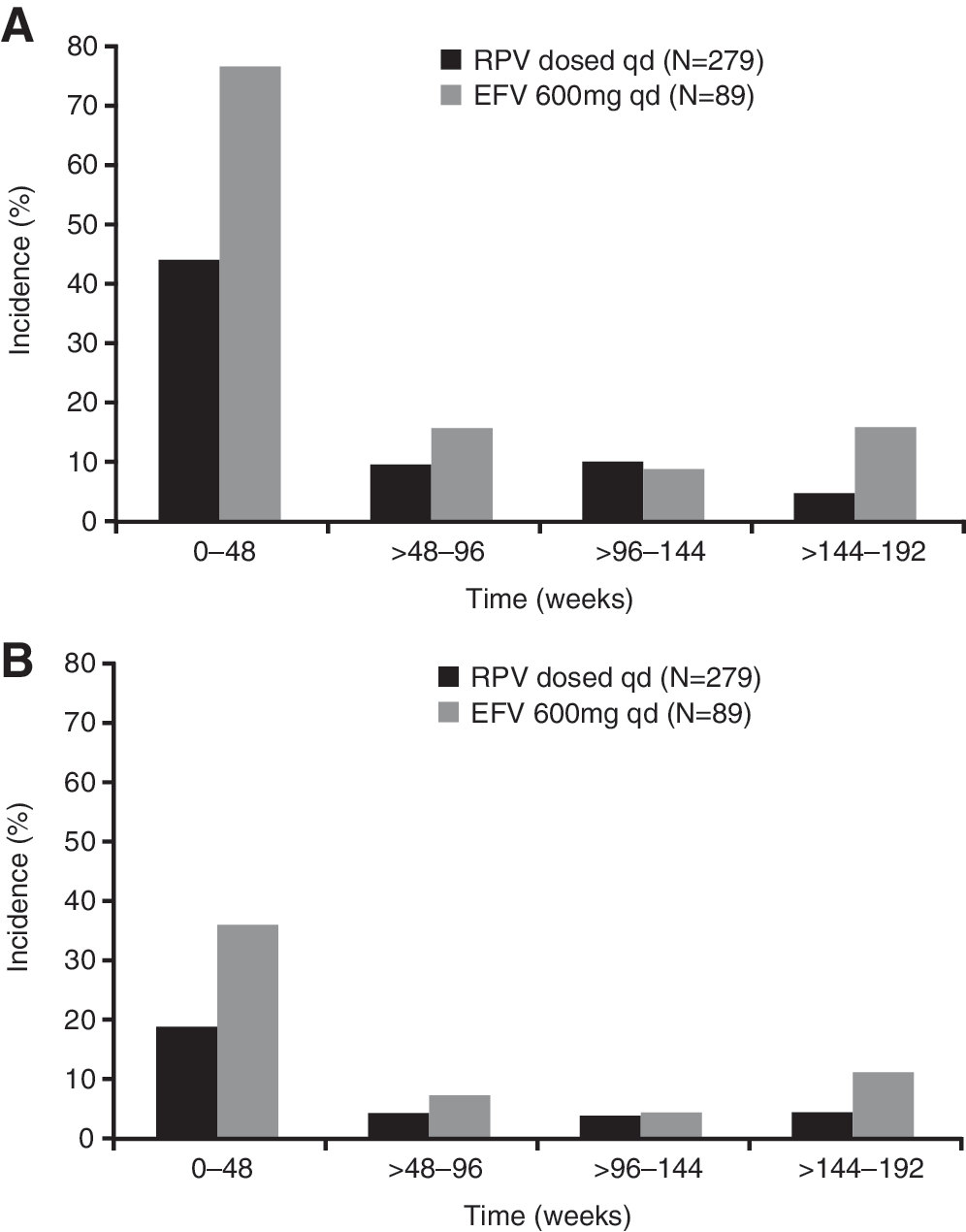
Incidence of any adverse event at least possibly related to RPV or EFV over time.
Excluding laboratory abnormalities reported as an AE.
Rash is defined as one of the following terms: allergic dermatitis, drug eruption, erythema, exanthem, rash, macular rash, maculopapular rash, papular rash, pustular rash, scaly rash, toxic skin eruption, urticaria, or papular urticaria.
Neurologic AEs are defined as one of the following terms: cluster headache, cranial neuropathy, disturbance in attention, dizziness, facial palsy, headache, lethargy, memory impairment, mononeuropathy, paraesthesia circumoral, photophobia, restlessness, sensation of pressure in ear, somnolence, uveitis, vertigo, or blurred vision.
Psychiatric AEs are defined as one of the following terms: abnormal dreams, affective disorder, aggression, agitation, anxiety, confusional state, depressed mood, depression, euphoric mood, homicidal ideation, insomnia, irritability, decreased libido, major depression, mood swings, nervousness, nightmare, panic attack, phobia, posttraumatic stress disorder, sleep disorder, social phobia, sopor, stress symptoms, or suicide attempt.
Treatment-emergent=worse than at baseline.
p<0.05; ** p<0.01; *** p<0.001 for EFV versus RPV; Fisher's exact test (post hoc analyses).
AE, adverse event; RPV, rilpivirine, EFV, efavirenz; N, number of patients per treatment group; n, number of patients with ≥1 event; aPTT, activated partial thromboplastin time; LDL-C, low-density lipoprotein-cholesterol; INR, international normalized ratio (measure of blood coagulation time).
The majority of rashes occurred early in treatment and resolved with continued dosing (median duration: RPV group 20 days versus EFV group 15 days). No grade 3 rashes or discontinuations due to rash were reported in the EFV or RPV groups beyond week 48, and no grade 4 rashes were reported. The majority of the neurologic AEs and psychiatric AEs were grade 1 or 2 and occurred predominantly during the first 48 weeks of treatment in both groups. In the week 192 analysis, incidences of grade 2–4 neurologic AEs regardless of causality were 7% in the RPV group versus 16% in the EFV group (p<0.05 RPV versus EFV) and incidences of psychiatric AEs were 9% versus 13%, respectively (p=0.32) (Table 3). Incidences of grade 2–4 neurologic AEs at least possibly related to RPV or EFV were 3% versus 9% (p=0.04) and incidences of psychiatric AEs were 2.5% versus 8% (p=0.05, all Fisher's exact test, post hoc analyses).
Similar incidences of serious AEs (16% versus 17%), grade 3 or 4 AEs at least possibly related to RPV or EFV (7% versus 9%), and AEs leading to treatment discontinuation (14% versus 12%) were reported in the RPV and EFV groups, respectively. After the week 48 analysis, only one serious AE considered at least possibly related to trial medication (inadequate control of diabetes mellitus) occurred in an RPV-treated patient (at week 104), while no additional serious AEs were reported in EFV-treated patients. After the week 96 analysis, a small proportion of RPV- (3%; 7/279) and EFV-treated patients (3%; 3/89) discontinued the trial due to an AE, mainly because of laboratory abnormalities reported as an AE and pregnancy, and two deaths occurred in the RPV group. One patient died at week 116 of an unknown cause, and one death at week 146 was a result of drug toxicity and intestinal infarction, both considered not related to RPV.
In the week 192 analysis, the overall incidence of treatment-emergent grade 3 or 4 laboratory abnormalities was similar for RPV and EFV (31% versus 29%, respectively) (Table 3). After an initial decline in hemoglobin levels in the RPV and EFV groups (noted primarily in the groups receiving the investigator-selected background N(t)RTI regimen of AZT/3TC) and recovery above baseline levels at week 96, hemoglobin levels continued to rise until week 144, reaching a plateau beyond this point. Mean [standard deviation (SD)] increases from baseline at week 192 in total cholesterol [17 (35) versus 41 (31) mg/dl, p<0.001 EFV versus RPV; nonparametric Wilcoxon rank-sum test, post hoc analysis] and low-density lipoprotein (LDL)-cholesterol [10 (30) versus 22 (31) mg/dl, p<0.01] were significantly lower in the RPV group than in the EFV group at week 192 (Fig. 4). Mean (SD) increases from baseline at week 192 in high-density lipoprotein-cholesterol (HDL-C) [5 (12) versus 11 (10) mg/dl, p<0.001] were also significantly lower for the RPV group at week 192 (Fig. 4), resulting in no difference in the change in ratio of total cholesterol/HDL-C between groups (−0.1 versus 0.002). There was a small increase from baseline in triglycerides for the EFV group, but not for the RPV group [10 (107) versus 54 (101) mg/dl, p<0.01] (Fig. 4).
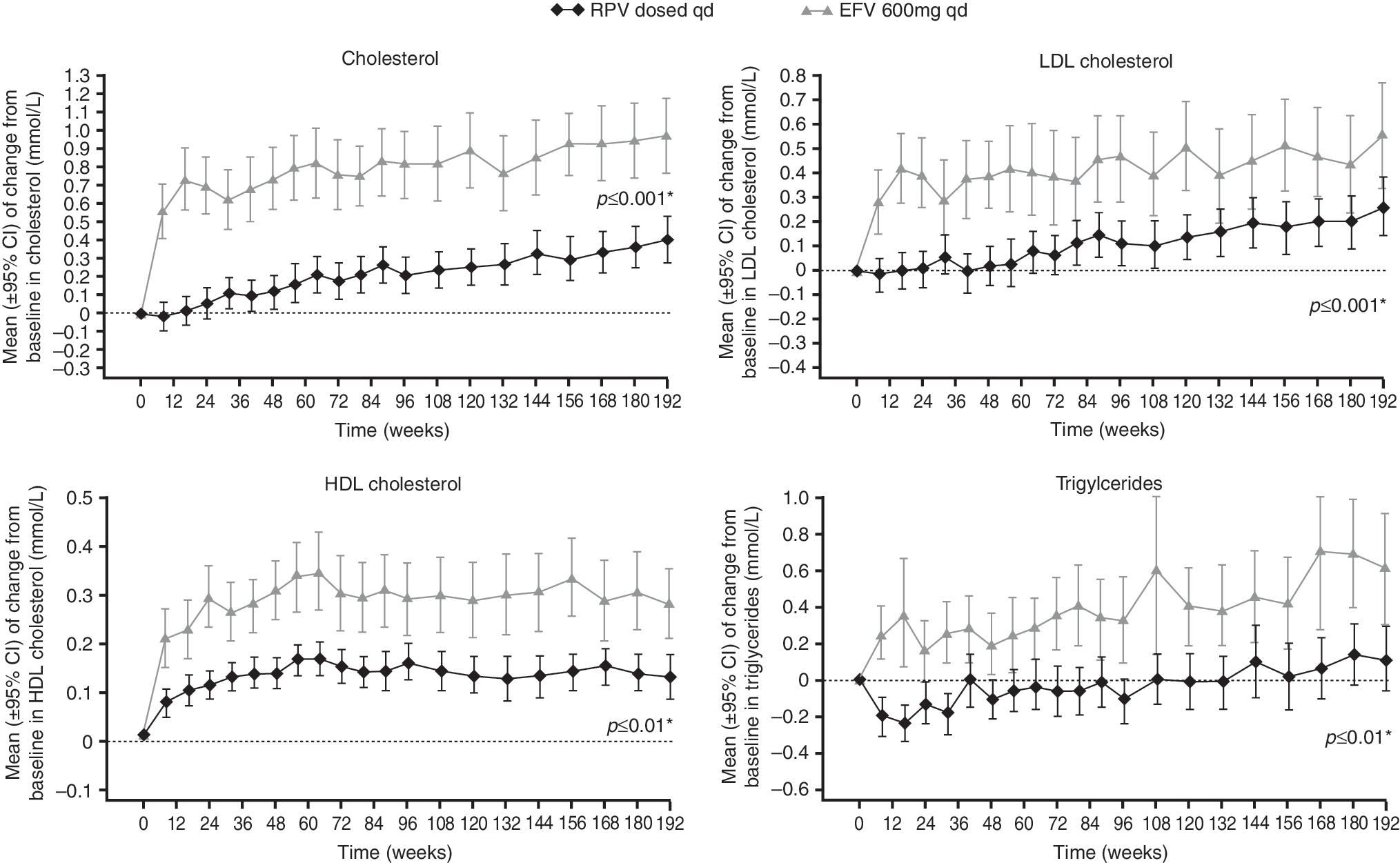
Mean change from baseline (standard deviation) in fasting lipid parameters over 192 weeks. *p value versus EFV at week 192 (nonparametric Wilcoxon rank-sum test; post hoc analyses).
In both treatment groups, mean change in QTcF interval observed up to week 48 (RPV group: 9.8 ms; EFV group: 12.0 ms) was followed by a further increase to week 192 (RPV: 16.4 ms; EFV: 14.4 ms). The proportions of patients with a treatment-emergent QTcF interval >450 ms were similar in each treatment group (7% versus 8%, respectively); none of these patients receiving RPV and one patient receiving EFV had a QTcF interval >500 ms. In addition, 10 patients receiving RPV and three patients receiving EFV (4% in each treatment group) had an increase in QTcF >60 ms from baseline. There were few AEs potentially related to QTcF interval prolongation (RPV 1% vs. EFV 2%).
Discussion
The objective of the extension to trial TMC278-C204 was to investigate the long-term safety, tolerability, and efficacy of RPV and compare these with data for EFV. There were no significant RPV dose-response relationships with respect to the primary endpoint (composite ITT-TLOVR algorithm) at week 48 or 96. 11 The RPV 75 mg qd dose was initially selected for phase III development since it allowed for drug–food or drug–drug interactions that could have lowered the exposure to that of the RPV 25 mg qd dose. The 25 mg qd dose of RPV was subsequently selected based on the results of the thorough QT studies and because the incidence of discontinuations due to AEs and rashes and increases in QTcF interval prolongation were lowest with this dose at week 96 in TMC278-C204. Given the lack of RPV dose-response for the primary endpoint, changes in RPV dose after week 96 were unlikely to have had an impact on trial outcome.
Once-daily oral RPV continued to show similar antiviral and immunologic efficacy to that of EFV over 192 weeks in HIV-1-infected, treatment-naive patients. In both groups, there were similar increases in mean change from baseline in CD4 cell count over time. The decrease in response rate from week 96 to week 192 was 10–14% in the treatment groups. This decline is in line with that observed in similar trials over 2–4 years of treatment, in which response rates have decreased by 5–10% each year. For example, data in the current trial lie in the same range as data from the GS903 and GS934 studies of EFV in treatment-naive patients in which 63–68% 15 and 58–71% 16 of EFV-treated patients, respectively, achieved a viral load <50 copies/ml at week 144 (ITT populations). In this study, there was no statistically significant difference in virologic failure rate between groups, with a 12% VFeff rate in the RPV group versus 8% for EFV (p=0.3), and the decline in TLOVR response rate over time was mainly driven by discontinuations for reasons other than virologic failure (Table 2). These included AEs leading to treatment discontinuation, loss to follow-up, withdrawal of consent, noncompliance, and completion of week 96, but noncontinuation into Extension 1. This is also consistent with previous observations in trials of treatment-naive patients. In a recent evaluation of 12 clinical trials of initial ARV therapy, the majority of treatment failures (73%) were due to discontinuation of study medication (32% for AEs and 41% for loss to follow-up) and only 27% were due to virologic failure. 17
The most frequent emerging NNRTI RAM in the RPV group was E138K and in the EFV group was K103N. In line with the background regimen containing FTC or 3TC, the most frequent emerging N(t)RTI RAM in the RPV group was M184V. In this trial M184V was not found in the EFV group. Given the small numbers of participants with resistance data at time of failure, no firm conclusions should be drawn from this trial regarding the resistance profile of RPV, yet observations made are consistent with the resistance profile of RPV seen in the Phase III trials. 8,9,18
RPV continued to be well tolerated through 192 weeks. Importantly, there were no new safety concerns, with no new types of AEs or notable increases in incidence of AEs between weeks 96 and 192. RPV was associated with a lower incidence of grade 2–4 AEs at least possibly related to trial medication than EFV. Incidences of rash and neurologic and psychiatric AEs often observed with EFV 2 occurred less frequently with RPV than with EFV in the week 192 analysis. EFV is also associated with metabolic changes, such as increases in cholesterol and triglycerides. 2 While increases in lipid parameters were higher with both NNRTIs at week 192 versus week 96 and versus baseline, 11 they remained significantly lower with RPV than with EFV. Treatment with RPV did not result in elevated triglycerides. There was no difference in the change in QTcF interval between the RPV and EFV groups, and the change is not considered clinically relevant given the low proportion of patients with prolonged QTc interval and AEs potentially related to QTcF interval prolongation in each treatment group. In a thorough QT trial in healthy volunteers, RPV 25 mg qd had no relevant effect on the QTcF interval. 14
Analysis of this trial extension has limitations because it was open-label and this may have influenced attribution of AEs. Additionally, the treatment groups were relatively small, particularly in EFV-treated group. In addition, the first trial extension at week 96 was not implemented in Uganda. This prevented further evaluation of these patients and to be conservative, patients from this site were counted as failures in the analysis irrespective of their response to treatment in the initial 96-week treatment period. Finally, there were two RPV dose switches, initially to 75 mg qd and then to 25 mg qd. However, it is unclear if this would have had any significant impact on the results since no relevant dose-response relationships for RPV at the primary endpoint were observed at week 48 or 96 (the completion of the dose-finding phase of the trial).
RPV demonstrated sustained antiviral efficacy and improved immunologic restoration that were similar to that seen with EFV over 192 weeks. This is consistent with the pooled 48-week analysis from two randomized, double-blind, Phase III trials in treatment-naive patients [ECHO (TMC278-C209); NCT00540449 and THRIVE (TMC278
Footnotes
Acknowledgments
We would like to thank the patients, investigators, staff, and trial coordinators from each center, and trial personnel from Tibotec Pharmaceuticals Ltd, Tibotec BVBA, Beerse, Belgium and Tibotec Inc., Titusville, NJ. This trial was sponsored by Tibotec Pharmaceuticals Ltd. We would like to acknowledge Eric Lefebvre, Guy De La Rosa, Ines Adriaenssen, David Anderson, Annemie Buelens, Sarah Fox, Stephan Marks, Gaston Picchio, Deborah Schaible, Kati Vandermeulen, Peter Williams, and Brian Woodfall for their input into this manuscript.
In addition to the authors, the TMC278-C204 study group included the following investigators and contributors: Argentina: Waldo Belloso, Pedro Cahn, Isabel Cassetti, Arnaldo Casiro, and Marcelo Losso; Austria: Armin Rieger and Norbert Vetter; Brazil: Clóvis Arns Da Cunha, Cláudio Gonsalez, José Valdez Madruga, Rogério de Jesus Pedro, and Artur Timerman; China: Li Xingwang and Hao Wu; France: Pierre-Marie Girard, Jean-Michel Molina, Dominique Salmon, Yazdan Yazdanpanah, and Patrick Yeni; Germany: Keikawus Arastéh, Gerd Fätkenheuer, Frank Goebel, and Joerg-Andres Rump; Russia: Boris Gruzdev, Oleg Kozyrev, Grigory Moshkovich, Alexander Pronin, Oleg Romanenko, Elena Vinogradova, and Alexey Yakovlev; South Africa: Prudence Ive, Steven Miller, Lerato Mohapi, and Robin Wood; Thailand: Ploenchan Chetchotisakd, Khuanchai Supparatpinyo, Wichai Techasathit, Asda Vibhagool, Anchalee Avihingsanon, and Chris Duncombe; Uganda: Elly Katabira; UK: Edmund Wilkins; United States: Nicholaos Bellos, Philippe Chiliade, Kunthavi Sathasivam, Charles Farthing, Jeffrey Nadler, Beata Casanas, Peter Shalit, Dewald Steyn, and Melanie Thompson.
Prior abstract publication describing parts of these data: Wilkin A, Pozniak AL, Morales-Ramirez J, et al.: TMC278 shows favorable tolerability and non-inferior efficacy compared to efavirenz over 192 weeks in HIV-1-infected treatment-naïve patients. 19th Annual Canadian Conference on HIV/AIDS Research, Saskatoon, Canada, 13–16 May 2010. Abstract 7214.
Author Disclosure Statement
The authors received medical writing support from Ian Woolveridge, Ph.D., of Gardiner-Caldwell Communications Ltd, Macclesfield, UK, which was funded by Tibotec.
Aimee Wilkin has received research funding from Tibotec, Pfizer, Merck Sharp & Dohme, Abbott Laboratories, Gilead Sciences, Bristol-Myers Squibb (co-investigator), and Schering-Plough (co-investigator) and served on advisory boards for Gilead Sciences, Merck Sharp & Dohme, and Schering-Plough. Beatriz Grinsztejn has participated in advisory boards, has received speaker fees and has been an investigator for clinical trials for Tibotec, Bristol-Myers Squibb, ViiV Healthcare, and Merck Sharp & Dohme. Kiat Ruxrungtham has received consultancy fees, or honoraria, travel grants, or research grants from Tibotec, F. Hoffmann-La Roche, Merck, Sharp & Dohme, Bristol-Myers Squibb, Gilead Sciences, Abbott Laboratories and GlaxoSmithKline and also has been partly supported by the Professional Researcher Strengthening Grant, the National Science and Technology Development Agency, BIOTEC, Minitry of Science and Technology, Thailand, the National Research University Project of CHE, and the Ratchadaphiseksomphot Endowment Fund (HR1161A). Anton Pozniak has received consultancy fees, and/or honoraria, travel grants, and/or research grants from Tibotec, F. Hoffmann-La Roche, Merck, Sharp & Dohme, Bristol-Myers Squibb, Gilead Sciences, Abbott Laboratories, GlaxoSmithKline, and Boehinger Ingelheim. Mario Santoscoy has attended advisory boards and/or received consultancy fees, and/or honoraria, and/or educational/travel grants from Abbott Laboratories, Aventis, Bayer, Boehringer Ingelheim/Promeco, Bristol-Myers Squibb, Gilead Sciences/Stendhal, GlaxoSmithKline, Merck Sharp & Dohme, Roche, Schering-Plough, Serono, Tibotec/Janssen-Cilag, and ViiV Healthcare. Laurence T. Rimsky, Simon Vanveggel, and Katia Boven are full-time employees of Tibotec.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.