
Abstract
HIV-2 group A is predominant in different parts of the world, especially Africa, Portugal, Spain, France, the United Kingdom, the United States, Korea, and India. Among the Asian countries, India accounts for about 95% of all HIV-2 infections. The prevalence of HIV-2 has been reported from various states of India such as Maharashtra, Goa, Tamil Nadu, West Bengal, and Uttar Pradesh. In the present study, we analyzed transmembrane region (gp36) sequences of 10 HIV-2 group A Indian strains, isolated from Indian HIV-2-seropositive individuals. HIV Blast analysis for the 1.0-Kb region of the gp36 transmembrane region has shown that all these sequences belong to HIV-2 group A. Phylogenetic analysis indicated that the sequences cluster with HIV-2 group A sequences of Cameroon and Senegal. The epitope found at position 645–656 (YELQKLNSWDVF), previously reported as a broadly neutralizing determinant, was very well conserved in all 10 study sequences. The percentage similarity between Indian and South African HIV-2 group A gp36 sequences was 90% (range 86–100, SD 2.8) and with other nonsubtype A clades was 84% (range 77–100, SD 6.06) indicating overall less variability among the reported HIV-2 sequences. Similarly, the consensus amino acid sequences of the envelope transmembrane region of HIV-1 (gp41) and HIV-2 (gp36) is quite synonymous, indicating 87% similarity; however, limited information is available about the gp36 transmembrane region of the prevalent HIV 2 group A Indian strain. The rate of synonymous substitutions reported in the gp105 region was significantly higher, suggesting lower virulence of HIV-2, which does translate into a lower rate of evolution, while the dN/dS ratio for the gp36 transmembrane region was less than one, indicating its conservation and significance (p<0.05) in structural and functional constraints.
Introduction
HIV-2,
The surface glycoproteins of enveloped viruses play a critical role in the initial events of viral infection, mediating virion attachment to cells and fusion of the viral and cellular membranes. The envelope glycoprotein of HIV-2, like that of other retroviruses, is synthesized as a polyprotein precursor (gp 160), which is proteolytically processed by a host protease to generate the surface envelope glycoprotein (SU gp105) and the transmembrane envelope glycoprotein (TM gp36). These two HIV-2 envelope glycoproteins are noncovalently associated subunits of gp160. The SU glycoprotein binds to the HIV receptor molecule CD4, while the TM glycoprotein anchors the SU/TM complex in the viral envelope or the plasma membrane of the infected cell. More precisely, the former gp105 directs target-cell recognition and viral tropism through interaction with the cell surface receptor CD4 and one of several coreceptors that are members of the chemokine receptor family, while the later transmembrane spanning the gp36 subunit promotes fusion of the viral and cellular membranes resulting in the release of viral contents into the host cell. 8 Moreover, the gp36 region has multiple functions ranging from membrane fusion, endocytosis signals, and calmodulin binding that potentially affect critical cellular signal transduction pathways. 9 These functions, along with structural elements that interact with the assembling capsid precursors, suggest that this region might play an important role in both virus replication and pathogenesis in vivo. 10
The full length genome of the HIV-2 virus, like other retroviruses, is highly variable and is subsequently classified into eight genetic subtypes. A study by Gao et al. reported that the average genetic divergence in the variable region of the envelope gene between different HIV-2 subtypes is about 20%. 11 In spite of such a divergence in the full length genome of HIV-2, we find 90% conservation in the gp36 region of the envelope gene, which appears to be important for antibody binding and an important target for neutralization. 12 To determine the genetic variability in the transmembrane region of the HIV-2 strain from western India, the gp36 region of the HIV-2 envelope gene from isolates of 10 HIV-2-seropositive individuals has been sequenced and analyzed.
Materials and Methods
The National AIDS Research Institute has developed a repository of Indian HIV strains that includes isolates of samples collected from 1996 to 2001. Ten heterosexually transmitted HIV-2 isolates (from individuals of ages ranging between 26 and 45 years) showing variability in the crucial functional domains were selected and the gp36 region was sequenced and analyzed. The study subjects were either symptomatic or asymptomatic with high or low CD4 counts. Blood specimens were screened for antibodies to HIV using HIV-1 and HIV-2 enzyme immunoassay (EIA) kits for anti-HIV-1 and anti-HIV-2 antibodies (Innotest HIV-1/HIV-2 Ab Immunogenetics, N.V. Zwijnarde, Belgium), followed by a differentiating rapid EIA (HIV TRIDOT Biotech Inc., Parwanoo, India) and confirmed as HIV-2 positive by Western blot (INNO-LIA HIV 1/II Score, Innogenetics, N.V. Technologiepark 6, Gent, Belgium).
The peripheral blood mononuclear cells (PBMCs) from the study subjects were separated and the viruses were isolated and confirmed as reported earlier. 13 Subsequently, the viral stocks were developed in phytohemagglutinin (PHA)-stimulated PBMCs and the cocultured cells were used for DNA extraction. About 6 μg of cellular DNA was extracted from 3×106 cocultured cells using the QIAamp DNA blood Mini kit (Qiagen, Hilden, Germany) according to the manufacturer's protocol. These HIV-2-positive DNA samples were used for amplification of the transmembrane region of the envelope (gp36) gene spanning the 1.0-Kb region successfully using an anchored polymerase chain reaction (PCR) technique. For amplification, the outer PCR primer pair was used as ST1: 5′ GGGGCTCGGGATATGKTATG 3′ [sense 6053–6072] and ST2: 5′ CAAGAGGCTTATCAGYTGGCGGATRCAGGAA 3′ [antisense 8414–8444] while the inner PCR primer pair was H2-7 5′ AGTTCTGMCACYTCTGCACT 3′ [sense 7862–7881] and ST2: 5′ CAAGAGGCTTATCAGYTGGCGGATRCAGGAA 3′ [antisense 8414–8444]. These outer and inner primer pairs were used for the first and second round of amplification, respectively. Following initial denaturation at 94°C for 2 min, cycling condition was set to 3 cycles of 94°C for 1 min for denaturation, 55°C for 1 min for annealing, and 72°C for 1 min for extension, followed by 32 cycles of denaturation at 94°C for 15 s, annealing at 55°C for 30 s, and extension at 72°C for 45 s with final extension for 5 min at 72°C. The Mg2+ concentration in both the rounds was 1.5 mM. Five microliters of the primary PCR product was used for the second round of reaction and amplified under PCR conditions similar to the first round using a primer pair H2-7 and ST2 except for annealing temperature as 58°C. Negative controls consisting of uninfected DNA and water were used in each run of the PCR reaction. An amplicon of 1.0 Kb size was generated, which was confirmed by running it on a horizontal gel electrophoresis (Fig. 1).
Horizontal gel electrophoresis (1% agarose) showing an amplicon of
After purification of the PCR product with a conventional ethanol precipitation method, these products were directly sequenced using a new panel of primers designed to cover the gp36 transmembrane region of HIV-2. This new panel of five primers was designed according to the HIV-2 consensus sequence using the Primer Express software (Applied Biosystems, Foster City, CA). The sequences of these primers are H2-8066 5′ GCTCAGTCTCGGACTTTA 3′ (nucleotide position 8066–8083), H2-8090 5′ GGGATAGTGCAGCAACAGCAA 3′ (nucleotide position 8090–8110), H2-8177 5′ CTCCAGGCAAGAGTCACTGC 3′ (nucleotide position 8177–8196), H2-8569 5′ GGCTATAGGCCTGTTTTCT 3′ (nucleotide position 8569–8588), and H2-8569RC 5′ GAGAAAACAGGCCTATAGCC 3′ (nucleotide position 8569–8588). These sequences were further resolved on an automated Genetic Analyzer ABI model 3100 (Applied Biosystems, Foster City, CA). The dN/dS ratio was calculated using the SNAP program (
Results
The demographic, epidemiological, and clinical details of the 10 study subjects are summarized in Table 1. The sequencing data revealed that all the sequencing primers generated around 500-bp fragments. These individual 500-bp fragments of each sample were assembled covering an overlapping region from both the ends and were used to generate a contig of 1.0 Kb and manually edited using the Lasergene software (DNASTAR Inc.) Version 5.05. The derived nucleotide sequences were aligned using the CLUSTAL X multiple sequence alignment program
14
with reference strains of HIV-2 subtypes A, B, AB, U, and G as well as SIV/MAC extracted from the Los Alamos Laboratory HIV sequence database (LANL) (
The nucleotide sequences of the gp36 transmembrane regions were translated using the Visual Sequence Editor [Vised] Version 1.1, 1996, by Ken Peters, Canada and then aligned by the Clustal X program. The online program

Alignment of HIV-2 gp36 sequences with the consensus sequence developed from 10 study participants. Different regions of gp36 sequences are indicated at the top of the consensus sequence. N-linked glycosylation sites are indicated by ^^^.
Phylogenetic analysis of study sequences together with reference sequences could determine the HIV-2 group classification or identify new groups if the sequences did not belong to any known group. The phylogenetic tree of study sequences indicated that all 10 subjects in this study were infected by HIV-2 group A and Indian sequences clustered with reference sequences from the Cameroon (CN), Guinea-Bissau (GW), and Senegal (SN) regions of West Africa, which was further supported in the neighbor-joining analysis by bootstrap value of 100 (Fig. 3). To further support the analysis of the phylogenetic tree, a graph of similarity versus position was plotted using SimPlot Version 3.5.1 software (provided by Stuart Ray) with a 200-nucleotide window and a step size of 20 to compare percent similarity among HIV-2 group A sequences from the current study samples with that of African HIV-2 group A sequences published from the Los Alamos database. The SimPlot indicated more than 85% similarity with published HIV-2 group A sequences along with no evidence of recombination (Fig. 4). The dN/dS ratio for our study samples is less than one (0.065) on comparing them with reported HIV-2 group A and representative sequences of different clades, strongly emphasizing the presence of sequence conservation at various disease stages and even across clades.
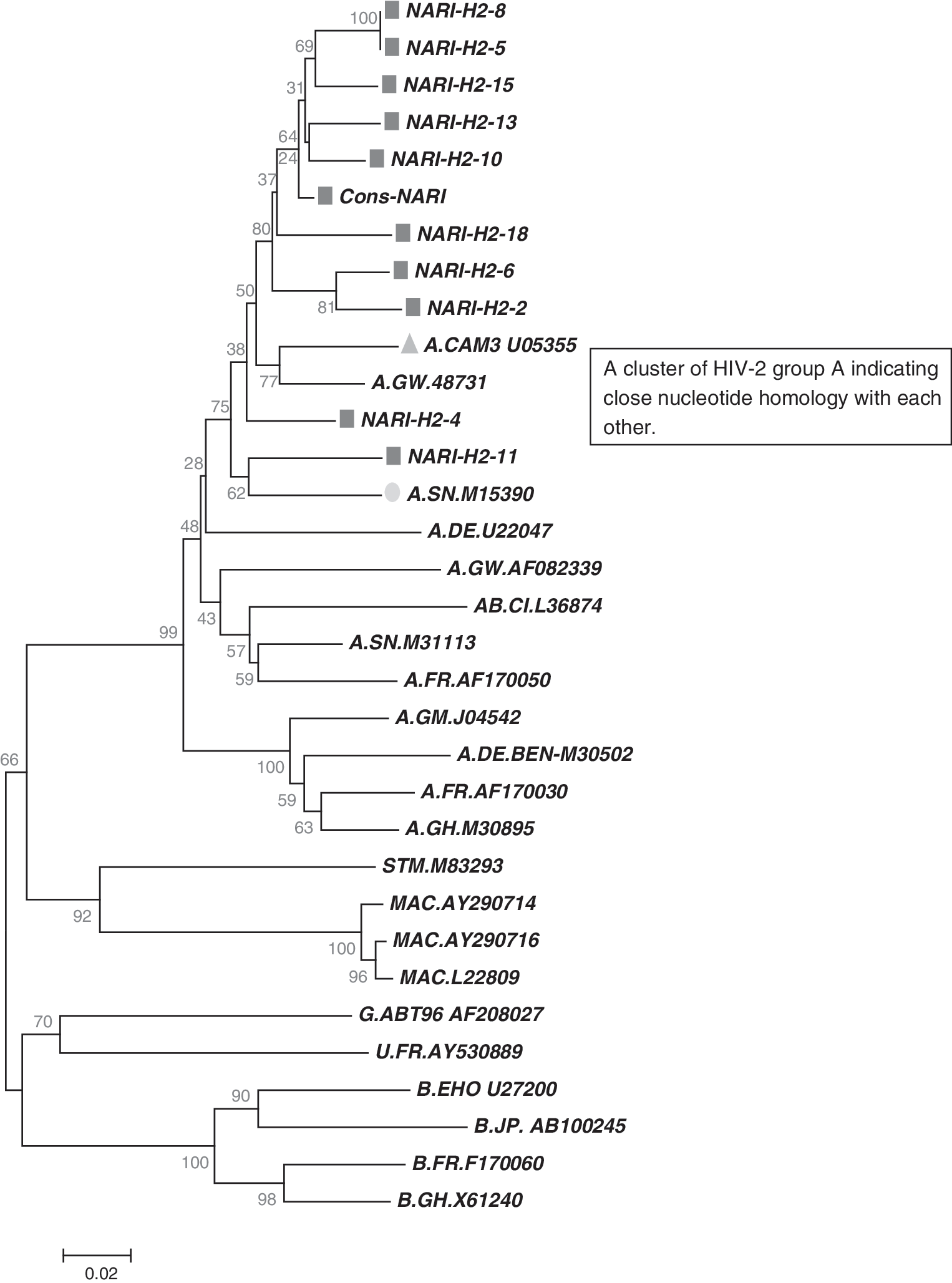
Phylogenetic analysis of gp36 sequences from 10 HIV-2 infected individuals. Reference sequences of all subtypes, SIVmac, SIVcpz were aligned using Clustal X software. The phylogenetic tree was constructed by MEGA 4.1. All the study sequences (square) found in single cluster along with HIV-2 group A sequences from Senegal (circle) and Cameroon (triangle). The scale of genetic distance is indicated may be printed.
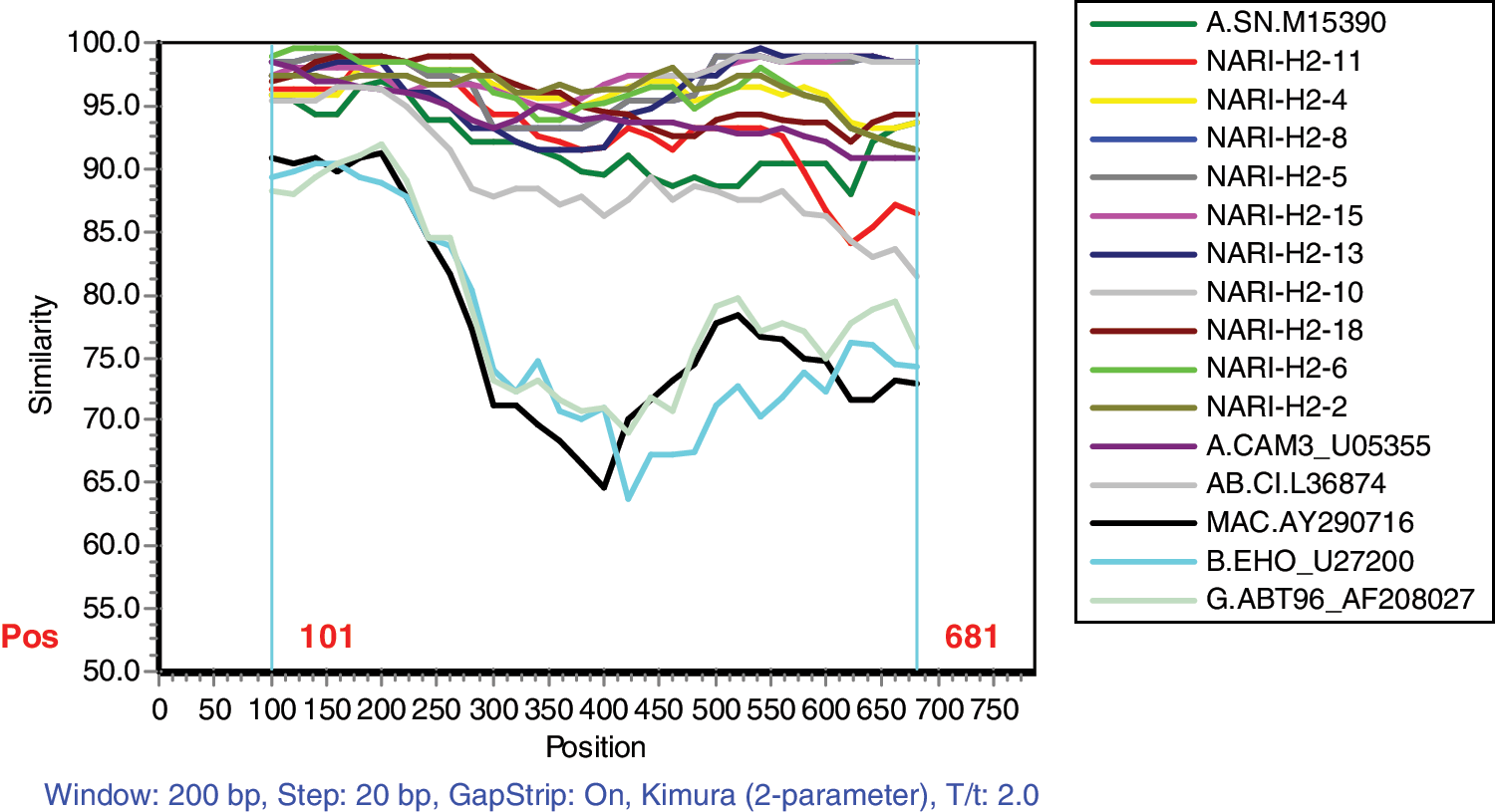
The SimPlot program was used to determine percentage similarity with respect to nucleotide sequences by comparing study sequences with reported subtype A, B, AB, G, MAC sequences from the Los Alamos database. Color images available online at
Discussion
Being highly immunodominant, several epitopes of HIV-2 have been suggested to harbor sites important for neutralization. 16 A functional study was carried out by Björling et al. in 199117 in which a panel of synthetic peptides was used for the evaluation of the neutralization response and antigenic activity in tests with human and simian sera. Measurement of neutralization and antibody-dependent cellular cytotoxicity (ADCC) had shown satisfactory performance against two, A5-25 [595–614 CHTTVPWVNDTLTPEWNNMT] and A5-28 [714–729 HIHKDWEQPDREETEE(C)], out of six synthetic peptides, A5-23 [573–595 AIEKYLADQARLNSWGCAFRQVC], A5-25 [595–614 CHTTVPWVNDTLTPEWNNMT], A15-82 [634–649 EQAQIQQEKNMYELQK(C)], A5-28 [714–729 HIHKDWEQPDREETEE(C)], A5-29 [724–739 EETEEDVGNDVGSRSW(C)], and A15-84 [830–845 LAVPRRIRQGAEIALL(C)]. However, after a period of more than 15 years, there seems to be significant variation in the amino acid sequences, which indicates that a new synthetic peptide panel needs to be developed for the currently circulating subtype-specific HIV-2 strain rather than following the same panel that had shown promising results in discriminating between infection with two different serotypes of the virus as well as results of high neutralizing antibody/low antigenicity and vice versa for those two, i.e., A5-25 [595–614 CHTTVPWVNDTLTPEWNNMT] and A5-28 [714–729 HIHKDWEQPDREETEE(C)] peptides. 17
Apart from this, epitope 645–656 (YELQKLNSWDVF) has been observed in all the sequences and has been reported as a broadly neutralizing determinant. 18 In contrast, the motif GYRPV (aa position 195–199) is present in the cytoplasmic tail (CPT) of HIV-2 and was conserved in our study sequences, which is responsible for enhancement of virus production. 19
Another functional study carried out by Lizeng et al. in 2003 21 mentioned for the first time that HIV-2specific serum IgA primarily bound to the 140–155 amino acid region of the transmembranous protein gp36 to which 72% of the sera reacted and neutralized a well-documented HIV-2SBL6669 strain of HIV-2. 20 Further purified IgA antibodies showed neutralizing effects against HIV-2SBL6669 in 17/29 cases (59%). Similar amino acid substitutions present in all current study sequences showed that there is a replacement of valine (position 152) with isoleucine and glycine (position 154) with serine. The overall charge of this peptide is not changed as valine and isoleucine both are nonpolar hydrophobic whereas glycine and serine are both polar uncharged. All the three N-linked glycosylation sites at the amino acid position of the study sequences (100, 109, and 125) (Fig. 2) respective to amino acid positions 603, 612, and 628 of the standard HIV-2 ROD sequence were well conserved across all study sequences. Furthermore, mapping of neutralizing antibodies to the carboxyl-terminal intracytoplasmic domain has been generated in guinea pigs, 16 although the relevance of such antibodies in vivo remains uncertain. 22
One more functional characteristic of HIV-2 envelope fusion and processing domains studied by Freed et al. in 1992 23 indicated that the hydrophobic amino terminus of the HIV-2 TM glycoprotein is a fusion domain. The substitution in the amino terminal hydrophobic region of the HIV-2 TM glycoprotein has completely abolished formation of syncytium indicating its role in the fusion process without affecting CD4 binding or envelope precursor processing. This fusion domain is conserved in all our study samples, indicating consistent syncytia formation during virus isolation.
Apart from this, the mechanism of dimerization of the envelope glycoprotein of HIV-2 is still unclear. The sequence analysis data could help us in identifying probable sites in this region, which would allow us to perform an immunological assay and to determine problems in the mechanism of dimerization of the glycoprotein precursor as well as homodimer formation during the processing of transmembrane envelope glycoproteins of HIV-2. 24
The phylogenetic analysis of gp36 has provided information that is useful for the determination of variants of HIV-2 that occur in the western part of India. The sequence analysis of HIV-2 has indicated and proven that this region of gp36 is also representative of the prevalence of HIV-2 subtype A in western India. The sequences presented here will be of immense help to researchers in designing small peptide analogues and in determining their function as therapeutic HIV-2 entry inhibitors. A substitution set of peptides representing the conserved antigenic site in the central part of gp36 will be helpful to identify the role of individual amino acids important for human antibody binding. Another important and urgent task is to determine the localization of neutralizing epitopes from the conserved gp36 region of HIV-2 because the virus appears to mutate to phenotypes that escape neutralizing antibodies. However, further surveillance of HIV-2 strains circulating in this region in other risk groups is required to determine whether any other HIV-2 group also exists in India.
Thus, using gp36 env region sequences for phylogenetic analysis of HIV-2 strains can provide an effective tool for the determination of distinct HIV-2 variants. In addition, an HIV-2 gp36 primer set may have a broad spectrum of future applications, ranging from quantitative measurement of viral load for clinical management of infected patients to its utilization as a diagnostic tool for early detection of HIV-2 infection in blood donors. Hence, gp36 sequences would provide valuable information regarding its conservation across various clades and its diagnostic implication within the distinct domains of the HIV-2 multifunctional protein.
GenBank Accession Numbers
HIV-2 transmembrane region (gp36) sequences presented in this study have been deposited in GenBank under accession numbers GQ338297–GQ338306.
Footnotes
Acknowledgments
The Virus Repository, from which the HIV-2 isolates were selected, was supported by the Department of Biotechnology (DBT), Government of India, and an intramural research grant from the India Council of Medical Research (ICMR), New Delhi. The author wishes to thank all those who have significantly contributed to generate the virus bank and to thank the director, NARI for allowing us to use the isolates available in the repository.
Author Disclosure Statement
No competing financial interests exist.