
Abstract
Significant subtype-specific differences were observed in the protease (PR) region of the HIV-1 pol gene. Most of the previous studies were restricted to subtype B, although subtype C accounts for more than 50% of HIV infections worldwide. In this study we characterized PR sequences from primary clinical isolates from protease inhibitor (PI)-naive patients in South India (n=39) as well as database-derived HIV-1 subtype C sequences from India (n=542) and globally (n=2970). All the study sequences were identified as subtype C, which is predominant in India. Drug resistance genotyping analysis identified 2.6% (1/39) prevalence of major PI resistance (I54T) and 7.7% (3/39) of minor PI resistance (L10I, T74S, and A71T). Selection of T12S, I15V, L19I, M36I, R41K, H69K, L89M, and I93L was observed both in global and Indian subtype C while the L63P mutation was selected in Indian PR sequences. Three different codon-based maximum likelihood methods agreed on four sites (12, 19, 36, and 82) under positive selection in Indian sequences.
I
HIV-1 subtype C, which was initially discovered in Ethiopia, 3 accounts for around 50% of all HIV infections worldwide and is highly predominant in the epidemics in Southern Africa and the Indian subcontinent. 4 Most of the research and drug design was carried out in subtype B, which is common in resource-rich settings. Data from non-B subtypes are inadequate and demand extensive research to address the drug resistance-related issues in these subtypes. Moreover, data on transmitted PI drug resistance in India are limited despite PIs being included as second-line antiretroviral therapy in the Indian National ART Program. In this study we sought to analyze the presence of transmitted PI resistance-associated mutations and naturally occurring polymorphisms due to viral natural evolution and host selection pressure exerted in the HIV-1 protease region. The sequences reported here include both primary clinical isolates (n=39) and database-derived sequences reported from India (n=542) and globally (n=2970). To our knowledge, this is the first study to simultaneously examine all available sequences from PI-naive HIV-infected individuals from India. We also compare the presence of naturally occurring polymorphisms within the HIV-1 subtype C PR region (1–99aa) of the pol gene with similar findings in other parts of the world.
Thirty-nine HIV-1-infected individuals attending the Infectious Disease Clinic at St. John's Medical College and Hospital, Bangalore, India who were previously unexposed to protease inhibitors were selected for the study. After obtaining informed consent, whole blood samples were collected from adult subjects (n=37) between April 2010 and February 2011. Two pediatric samples were collected in 2007. All 39 subjects were diagnosed with HIV infection using three separate rapid antibody assays as per the National AIDS Control Organization (NACO, Ministry of Health and Family Welfare, Government of India) guidelines. This study was approved by the Institutional Ethical Review Board of St. John's Medical College and Hospital. A routine CD4 count was performed using a dual-platform flow cytometer (FACSCalibur, BD, USA). Viral load was determined by an Abbott m2000rt RealTime HIV-1 assay in an m2000rt machine (Abbott Molecular Inc., USA) as per the manufacturer's directions.
One milliliter of plasma was centrifuged at 4°C for 1 h at 14,000 rpm. Viral RNA was extracted from 140 μl of concentrated plasma using a commercial kit (QIAamp viral RNA extraction kit, Qiagen, Germany) and reverse transcribed to complementary DNA (cDNA) with random hexamer primers (Promega Corporation, USA). The HIV-1 protease region (1–99 amino acids, HXB2 position 2253 to 2550) from cDNA was amplified using a seminested in-house polymerase chain reaction method using iNtRON Taq polymerase (Intron Biotech, South Korea). The primers have been designed based on the Indian subtype C consensus sequences created using 166 previously reported sequences from India. First round PCR was performed with primers UNPR1F- 5′-TTTTAGGGAAAATTTGGCCTTCC-3′ (HXB2 position 2087 to 2109) and UNPR1R 5′-TTCTTTTTTATRGCAAATATTGGAG-3′ (HXB2 position 2722 to 2746) with the cycling profile 94°C for 5 min [94°C 20 s, 55 °C 45 s,72 °C 1 min 20 s]×40 cycles and 72°C for 7 min. Second round of PCR was conducted using UNPR1F and UNPR2R-5′-GGATTTTCAGGCCCAATTTTTG-3′ (HXB2 position 2691 to 2713) with the cycling profile 94°C for 94 5 min [95°C 20 s, 50°C 30 s, 72°C 45 s]×35 cycles, 72°C for 7 min. Column purified (QIAquick Gel Extraction Kit, Qiagen, Germany) nested PR amplicons were bidirectionally sequenced using UNPR1F and UNPR2R primers in a 3730xl DNA analyzer (Applied Biosystems, CA). Sequences were manually edited using BioEdit sequence alignment editor version 7.0.5.3 and a consensus sequence was created.
Subtyping was carried out using three different approaches as described previously.
5
Multiple sequence alignment was carried out in Clustal W (
Selection pressures on proteins are quantified by the ratio of substitution rates at nonsynonymous (dN) and synonymous sites (dS) (dN/dS ratio). dN/dS <1 indicates purifying selection while dN/dS >1 indicates positive selection. Three different codon-based maximum likelihood methods were used to estimate the dN/dS ratio at every codon position in the alignment to determine if positive selection occurred. These methods included (1) single-likelihood ancestor counting (SLAC), which employs the number of dN and dS changes that occurred at each codon throughout the evolutionary history of the sample, (2) fixed effects likelihood (FEL), which directly estimates dN and dS substitution rates at each site, and (3) random effects likelihood (REL), which models variation in dN and dS substitution rates across sites according to a predefined distribution with the selection pressure at an individual site inferred using an empirical Bayers approach. 7 We have used all three methods as recommended previously to rule out spurious results. 7
All statistical analysis was carried out in Microsoft Excel 2007 (Microsoft Corporation, USA) and GraphPad QuickCalcs (
The cohort description was as follows: of 39 patients enrolled 27 (69%) were male. The median age was 34 years (range 10–57), the median CD4 count at the time of study was 241 cells/mm3 (range 8–616), and the median viral load was log10 5.42 copies/ml (range 3.18–6.47). Among the patients, the likely route of transmission for 36 (92.3%) was heterosexual transmission followed by 2 (5.1%) vertical transmission and 1 (2.6%) blood transfusion.
Phylogenetic analyses of the study sequences are shown in Fig. 1. All study sequences showed monophyletic branching with reference Indian HIV-1 subtype C strains, implying that all the strains were HIV-1 subtype C based on the protease gene, which is known to be predominant in India. All the study sequences demonstrated well-separated branches indicating the absence of cross-contamination. Subtyping using online subtyping tools (REGA subtyping tools, NCBI viral genotyping tools, and RIP 3) also confers similar results.
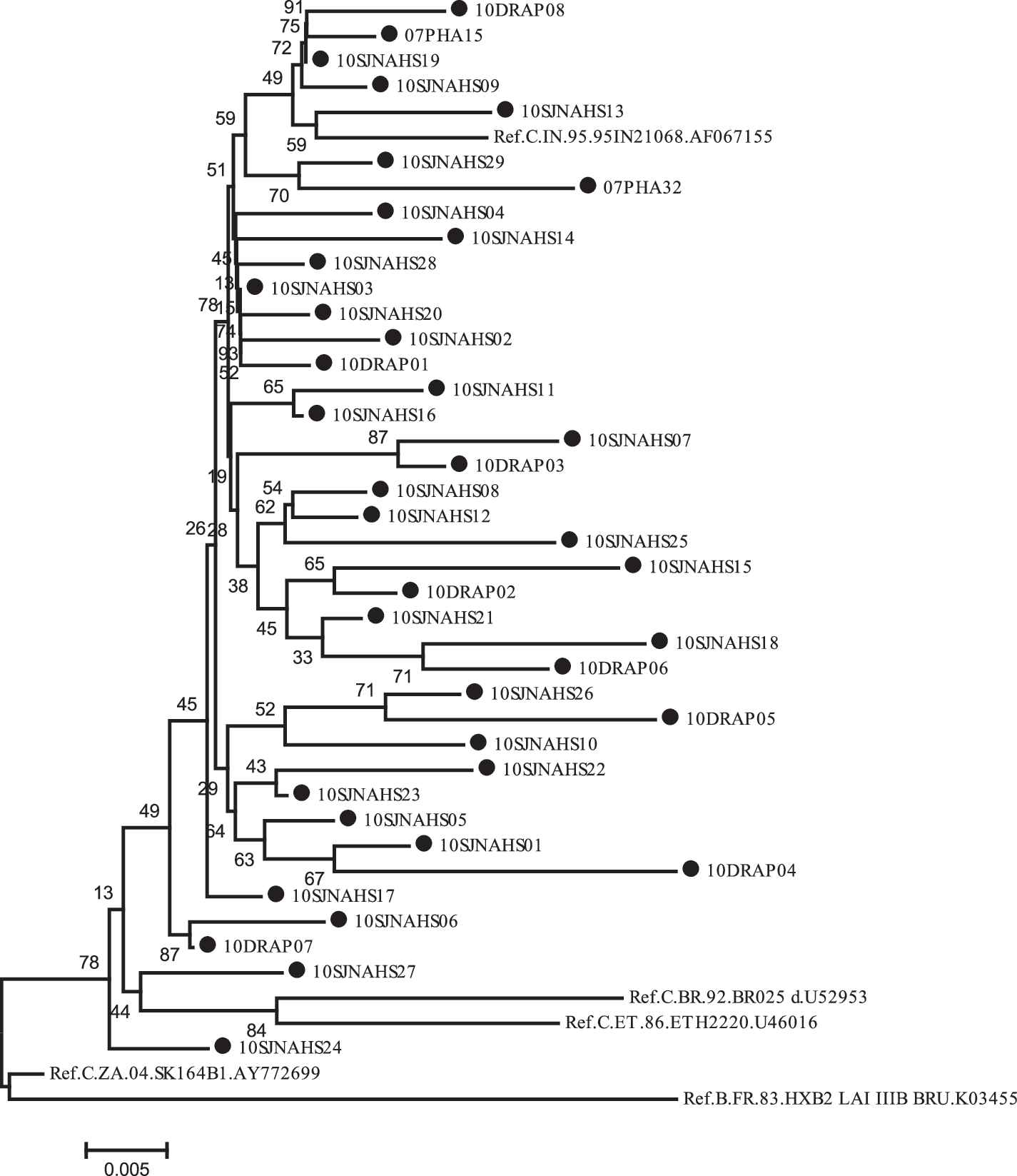
Phylogenetic analysis of the protease region of the HIV-1 pol gene (2253 to 2550 of HXB2 coordinate, 1–99 aa) of reference subtype C sequences and study isolates with HXB2 sequences as outlier. All study sequences (marked with a filled circle) were seen with well-separated monophyletic branching compared to reference Indian HIV-1 subtype C strains. None of the strains was identical, thus precluding cross-contamination.
Drug resistance genotyping analysis identified 2.6% (1/39) of isolates as having major PI resistance (I54T) and 7.7% (3/39) as minor PI resistance (L10I, T74S, and A71T). I54T confers low-level resistance against most PIs except darunavir/r. L10I is associated with resistance to most PIs when present with other mutations and is also present in 5–10% of untreated persons. T74S is associated with reduced nelfinavir susceptibility and A71T occurs in 2–3% in untreated persons (Stanford University HIV drug resistance database;
The comparative frequency of mutations in primary clinical isolates (n=39) with global subtype C sequences (n=3512) from PI-naive patients is given in Fig. 2A. There are 24 positions where >5% polymorphisms from wild-type consensus B sequences were observed either in cohort sequences or in global subtype C sequences. Significant differences between previously reported subtype C sequences and cohort sequences had been observed at positions T12, I13, G16, K20, R41, L63, I64, V82, and L89 (Chi square test; p<0.05). Multiple sequence analysis of consensus B and C and Indian C sequences indicated that 8% of the protease positions differ between Con_B and Con_C (T12S, I15V, L19I, M36I, R40K, H69K, L88M, and I93L) and 9% between Con_B and Con_C_IN (T12S, I15V, L19I, M36I, R41K, L63P, H69K, L89M, and I93L) (Fig. 2B). The L63P mutation has been selected in Indian PR sequences. We further assessed the frequency of L63P mutations in 581 Indian subtype C sequences (542 database derived and 39 primary clinical isolates) and 2970 global subtype C sequences (excluding Indian C sequences) and observed a significantly higher prevalence among Indian sequences (63.2 vs. 30.2; p<0.0001; Fig. 2C).
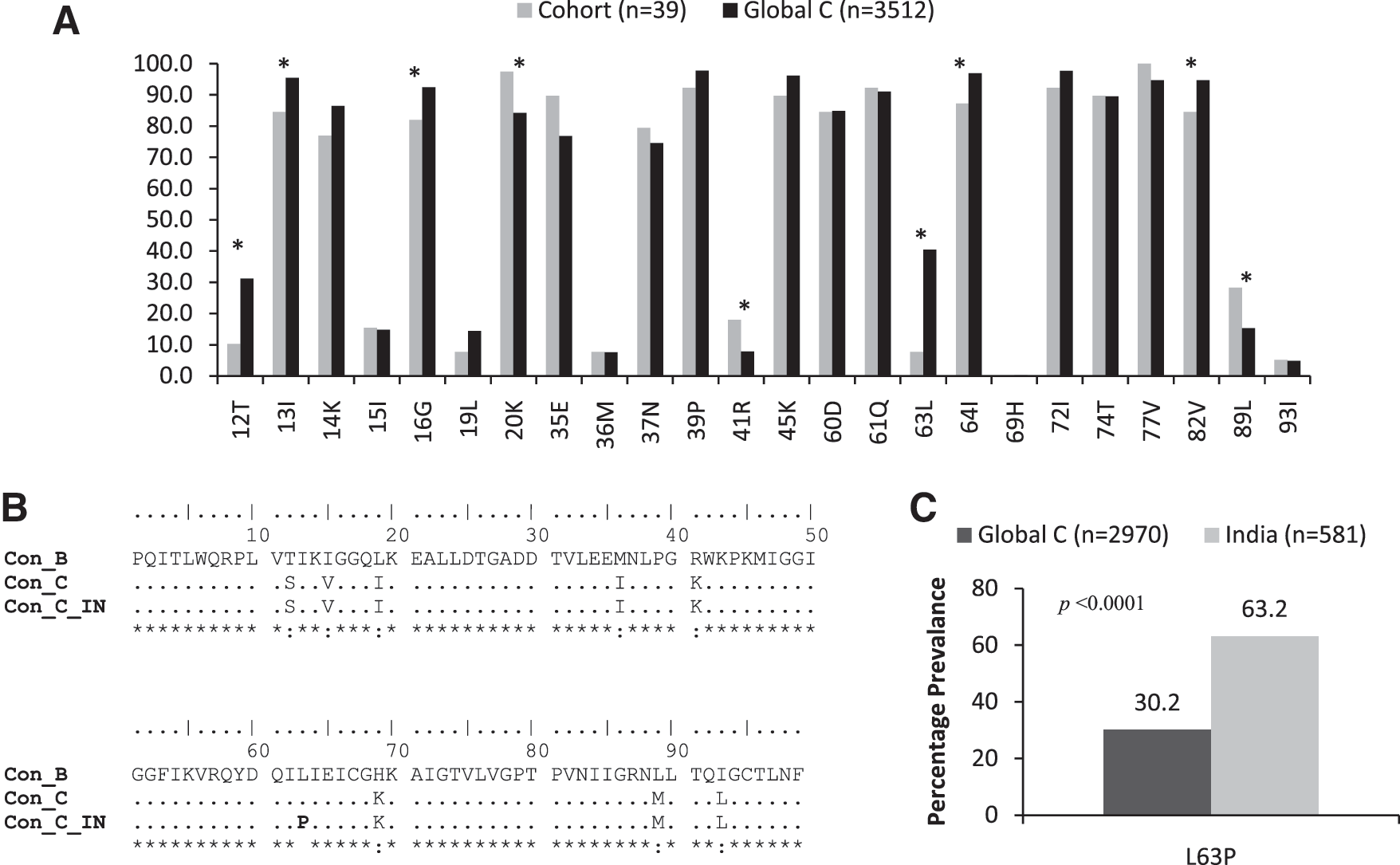
Polymorphisms in HIV-1 subtype C sequences with respect to consensus B sequences.
The alignment wide dN/dS ratio in Indian sequences was estimated at 0.24, which was concordant with the previous findings for selection in HIV-1 PR. 8,9 All three types of methods (SLAC, FEL, and REL) agreed on four sites (12, 19, 36, and 82) under positive selection (Table 1) and among them 12, 19, and 36 were selected for subtype C (Fig 2A). Two of the three methods agreed with another two sites, 15 (SLAC and REL) and 74 (SLAC and FEL), under positive selection. Site 15 was also selected for subtype C sequences. A single site, 37, was indicated to be a positively selected site by the REL method.
The dN/dS ratio is shown for each method. p-values for each appropriate tests and the Bayers factor values and posterior probabilities for REL methods tests are shown in parentheses. All three classes of methods (SLAC, FEL, and REL) agreed on four sites (12, 19, 36, and 82) under positive selection in Indian strains.
SLAC, single-likelihood ancestor counting; FEL, fixed effects likelihood; REL, random effects likelihood.
In this study we characterized all available database-derived PR sequences from PI-naive patients from India. A low baseline prevalence of PI transmitted drug resistance mutations was observed in our population. The overall prevalence of major PI drug resistance mutations in our cohort study was 2.6%, which was concordant with previous findings from India. 10,11 A report from South India concluded that 100% of minor PI mutations and 20% of major PI mutations were found in PI-naive individual based on the algorithm developed for clade B. 12 This may not reflect true cases because subtype C protease has several polymorphisms that are considered to be drug resistance mutation positions in clade B (e.g., L63P, which has been selected in Indian subtype C sequences).
The high conservation of protease residue found in Indian PR (dN/dS=0.24) likely reflects the sluggish evolution and molecular adaptation of protease function in Indian subtype C-infected individuals. Among the eight subtype C selective mutations 50% are found to be positively selected (Positions 12, 15, 19, and 36) by at least two methods of analyzing selection pressure. Previous studies have reported that mutated M36I viruses have a higher replication capacity than wild-type subtype B viruses. 13
In summary, this study provides the largest analysis of subtype C protease sequences from India. Polymorphisms at position M36I, R41K, H69K, and I93L were most frequent in both Indian and global subtype C sequences from PI-naive patients and most likely represent the natural genotypic variants in subtype C sequences. Additionally, L63P was selected in Indian sequences. The R41K and H69K polymorphisms are most likely accountable for CTL recognition of known epitopes in the Indian population as previously observed in CRF01_AE-infected patients in Southeast Asia. 9 Taken together, we may conclude that protease catalytic activity and the response to PI therapy may differ in different subtypes, and that resistance-associated mutations in subtype B may not necessarily translate to resistance in other subtypes. It is therefore wise to exert caution while interpreting PI drug resistant-associated mutations present within non-B subtypes to avoid erroneous prediction of drug resistance for non-B HIV-1 subtypes.
Footnotes
Acknowledgments
The authors would like to thank the Infectious Disease Clinic staff for their generous help with field work and sample and data collections. We would also like to thank our study participants who so generously contributed toward our understanding of issues in drug resistance genotyping in this setting. This study was partially funded by an EU FP7 grant. Sequences generated from this study have been submitted to GenBank (accession numbers JF451143–JF451181).
Author Disclosure Statement
No competing financial interests exist.