
Abstract
The C868T single nucleotide polymorphism in the CD4 receptor encodes an amino acid substitution of tryptophan for arginine in the third domain. Previous studies suggest that C868T increases the risk of HIV-1 acquisition; however, the influence of this single nucleotide polymorphism (SNP) on disease progression has not been established. The presence of the C868T polymorphism was not statistically significantly associated with HIV-1 disease progression outcomes in a cohort of postpartum Kenyan women.
Introduction
R
Only five nonsynonymous single nucleotide polymorphisms (SNP) are in the CD4 receptor, the primary receptor for HIV-1. The C868T (rs28919570) SNP in the CD4 receptor results in an amino acid substitution of tryptophan for arginine in the third domain. 9 Even though gp120 binds with the D1 portion of the CD4 receptor, this single amino acid change in the D3 region may modify the tertiary structure of the CD4 receptor, altering the interactions among the CD4 receptor, chemokine receptors, and gp120. One hypothesis is that this single amino acid change in the CD4 receptor leads to changes in the strength of binding to gp120, and subsequently altered risk in HIV-1 acquisition and HIV-1 disease progression.
In support of this, a previous study demonstrated that Kenyan infants with the 868T allele were more likely than wild-type infants to acquire HIV-1. 10 In addition, C868T was associated with an increased incidence of HIV-1 infection in Kenyan female commercial sex workers. 11 While these two studies suggest that C868T influences the risk of HIV-1 acquisition, the influence of this SNP on disease progression has not been established.
This study was designed to determine whether the C868T SNP was associated with increased risk of HIV-1 disease progression in a cohort of postpartum women in Kenya. Disease progression was defined using the following variables: death, reduction in CD4 cell count below 200 and below 350 cells/μl, rate of decrease in CD4 cell count over time, and rate of increase in HIV-1 RNA plasma levels over time.
Materials and Methods
Study setting and subjects
HIV-1-seropositive pregnant women were recruited from antenatal clinics in Nairobi and provided written informed consents. This study received ethical approval from the Institutional Review Boards of the University of Washington, University of Manitoba, and the Kenyatta National Hospital. Study participants were enrolled at 32 weeks gestation and began taking zidovudine twice daily between 34 and 36 weeks of pregnancy and continued through delivery following Kenyan national guidelines at the time of the study. 12
Women were seen in the clinic antenatally, at delivery, 2 weeks after birth, and then monthly for 12–24 months. Maternal blood specimens were collected at month 1, 3, 6, 9, 12, 18, and 24 months after delivery for CD4 cell count and HIV-1 RNA viral load (VL).
Laboratory procedures
HIV-1 RNA VL was quantified in plasma using the Gen-Probe Transcription Mediated Amplification assay, which is sensitive for detection of Kenyan HIV‐1 subtypes A, C, and D (Gen-Probe Incorporated, San Diego, CA). 13,14
Genotyping was performed by sequencing analysis, as previously described. 11 Maternal DNA was extracted from plasma using the AIAamp DNA Mini Kit. CD4 DNA was then amplified using a nested PCR protocol (due to low DNA yield) with two sets of primers: outer amplifying primers 5-GTCCAGGAATCCTAAGGACAGC-3 and 5-CCACCAGGTTCACTTCCTGATG-3 and inner amplifying primers 5-GTGGCCTGCTGTAGGAAAATGC-3 and 5-CACCAGGTTCACTTCCTGATGC-3. The PCR product (50 μl) was then purified using Montage PCR Centrifugal Filter Devices (Millipore) according to manufacturer's protocol. The purified product was then cycle sequenced with the following primers: 5-GGTAGGAAGGAACTGAAGTATCTG-3 and 5-TTCCTGTTTTCGCTTCAAG-3. Genotypes were determined by manual inspection of the SNP locus.
Statistical analysis
Kaplan–Meier survival analysis and Cox proportional hazard regression models were used to compare mothers with and without CD4 polymorphism for the following time dependent outcomes: maternal death, CD4<200, CD4<350, CD4<200 or death, and CD4<350 or death. These models were adjusted for baseline CD4 (postpartum month 1). Participants were censored at the last CD4 count measure. In addition, women who started HAART or had a second pregnancy were censored. Linear mixed effects models were used to determine associations between the CD4 polymorphism and rate of change in CD4 and VL over time, adjusting for baseline CD4 and baseline VL, respectively. In these models, mothers with fewer than two recorded CD4 cell counts or VL, respectively, were excluded from the analyses. Linear regression was used to compare maternal baseline CD4 cell count and baseline log10 transformed plasma HIV-1 VL between mothers with and without the C868T polymorphism. Analyses were performed using maternal genotypes in two models: (1) a dominant model, which assumed that risk was the same in mothers with either one or two T alleles; and (2) a codominant (allele–dose) model, which assumed that there was a constant change in risk with the number of T alleles. All analyses were specified a priori and data were analyzed in STATA version 11 (College Station, TX).
Results
Maternal polymorphism data were available for 449 mothers: 338 (75%) were wild-type (C/C), 88 (19%) heterozygotes (C/T), and 23 (5%) homozygous variants (T/T). The genotype frequencies (C allele=85%, T allele=15%) in this cohort differed from the Hardy–Weinberg equilibrium, using a chi-square test. Among these mothers, the median age, maternal CD4 count at 1 month postpartum, and plasma HIV-1 VL at 1 month postpartum were 25 years, 540 cell/μl, and 4.79 log10 copies/ml, respectively. Age, parity, marital status, maternal CD4 count at 1 month postpartum, and plasma HIV-1 VL at 1 month postpartum did not significantly differ between women with and without the T allele (Table 1). In addition, among 438 mothers, 15 (3%) died during 24 months of follow-up (Table 1), and no association was found between the C868T polymorphism and death (Table 2). Of the 15 death, 13 deaths were HIV related, 1 death was non-HIV related, and 1 death was due to unknown causes.
Adjusted for baseline CD4 count.
CI, confidence interval.
In both unadjusted and adjusted analyses, there was no statistically significant association between the C868T polymorphism and the four outcomes of interest: CD4 cell count below 200 and below 350 alone and combining death and CD4 cell count decline (Table 3). However, a trend toward a slower CD4 cell count decline below 200 [hazard ratio (HR): 0.45; confidence interval (CI): 0.20, 1.02; p=0.06] and CD4 cell count decline below 200 or death (HR: 0.50; CI: 0.23, 1.09; p=0.08) was seen in heterozygous and homozygous mothers compared to wild-type mothers when adjusted for baseline CD4 cell count.
Cumulative event rate by 2 years.
Adjusted for baseline CD4 count.
HR, hazard ratio; CI, confidence interval.
In the unadjusted analysis, on average, wild-type mothers lost 8.34 CD4 cells per month, while heterozygous or homozygous mothers lost CD4 cells at an average rate of 5.65 cells per month (p=0.17). Wild-type mothers lost 2.69 more CD4 cells per month than mothers with the T allele (Fig. 1). When adjusted for baseline CD4 cell count, there was trend for the 868T polymorphism to be associated with less rapid loss in CD4 cell counts over time. On average, wild-type mothers lost 8.80 CD4 cells per month, while heterozygous or homozygous mothers lost CD4 cells at an average rate of 5.01 cells per month (p=0.09).
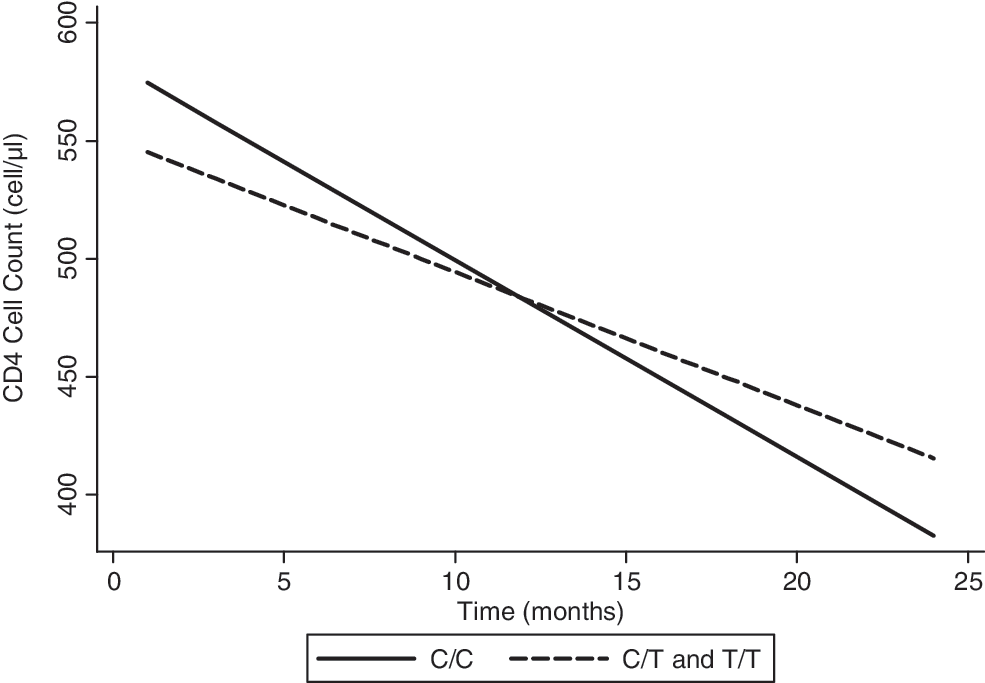
Expected CD4+T cell loss over time by C868T polymorphism, as predicted from a linear mixed effects model, unadjusted. On average, wild-type mothers lost 8.34 CD4 cells per month, while heterozygous or homozygous mothers lost CD4 cells at an average rate of 5.65 cells per month (p=0.17).
In both unadjusted and adjusted analyses, no association was found between the C868T polymorphism and viral load at baseline and over time (Fig. 2). Results were presented only in the dominant model because the models had similar findings.
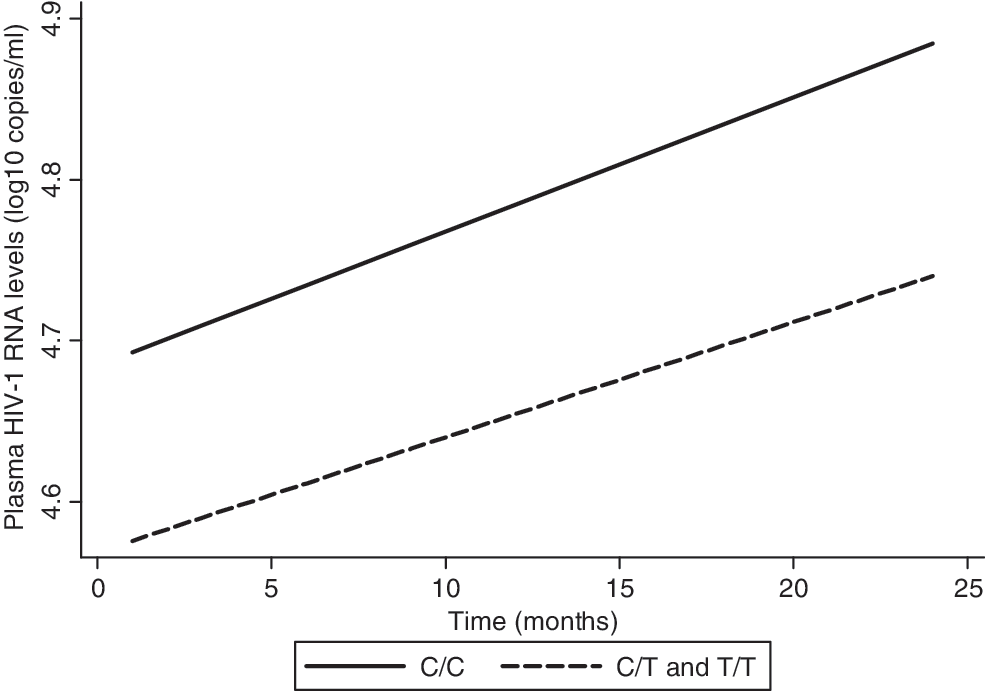
Expected HIV-1 viral load increase over time by C868T polymorphism, as predicted from a linear mixed effects model, unadjusted. On average, wild-type mothers gained 0.0083 log10 copies of HIV-1/ml per month, while heterozygous or homozygous mothers gained at an average rate of 0.0071 log10 copies of HIV-1/ml per month (p=0.88).
Discussion
The presence of the C868T polymorphism was not associated with HIV-1 disease progression outcomes in this cohort of postpartum Kenyan women. Rather than an increase in progression as we had hypothesized, we observed a trend toward a slower CD4 cell count decline over time in heterozygous and homozygous mothers compared to wild-type mothers when adjusting for baseline CD4 cell count. In addition, we observed a trend that showed heterozygous and homozygous TT mothers were less likely to develop CD4 cell count below 200 and CD4 cell count below 200 or death over time when adjusted for baseline CD4 cell count.
To our knowledge, there are no reports in the literature describing an association between the C868T SNP and HIV-1 disease progression. Past studies have shown increased risk of acquiring HIV-1 in infants and female commercial sex workers 10,11 and this led us to hypothesize that the presence of the C868T polymorphism would increase disease progression. One possible explanation for our finding the inverse is that the C868T SNP acts differently in the uninfected compared to HIV-1-infected individuals. Altered binding affinity, the hypothesized mechanism for higher risk of acquisition in HIV-1-uninfected patients, may be associated with decreased risk of HIV-1 disease progression. Since the C868T SNP involves a CD4 cell receptor, an essential immune molecule for HIV-1, it is possible that this SNP affects immune function by counterbalancing the increased risk of acquisition of HIV infection.
Another explanation is that other genetic immune factors and SNPs in linkage disequilibrium have a greater influence on HIV-1 disease progression than the C868T polymorphism. For example, the presence of HLA-B27 15 and HLA-B57 8,16 –18 alleles has been shown to be associated with delayed HIV-1 disease progression, and we were not able to assess the effect of HLA because typing was unavailable. The presence of these HLA alleles in our cohort may have nullified the impact of C868T on HIV-1 disease progression. It is also possible that other SNPs in linkage disequilibrium with C868T had an effect on HIV-1 disease progression, although no SNPs that we know of have been reported.
In summary, we found that mothers with the 868T allele did not have an association with increased HIV-1 disease progression. However, mothers with the 868T allele showed a trend toward delayed HIV-1 disease progression when adjusted for baseline CD4. The mechanism for this protective trend in HIV-1 disease progression is unknown, and more research is necessary to define the potentially complex interaction of the C868T polymorphism and HIV-1. Improved understanding of the impact of this polymorphism and other SNPs on the CD4 cell receptor may contribute to novel therapeutic strategies to alter HIV-1 binding affinity to decrease disease progression.
Footnotes
Acknowledgments
This research was supported by NICHD Grant HD-23412 and Canadian Institutes for Health Research (CIHR) Grants MOP-81377, HOP-75348, and MDP-86721. Additional funding was provided by the University of Washington's AIDS International Training and Research Program (AITRP) supported by the Fogarty International Center, National Institutes of Health (D43 TW000007). R. Choi was supported by an International Research Scientist Development Award (K01 TW008406). Also, R. Choi was supported by the National Institutes of Health Office of the Director, Fogarty International Center, Office of AIDS Research, National Cancer Center, National Eye Institute, National Heart, Blood, and Lung Institute, National Institute of Dental & Craniofacial Research, National Institute on Drug Abuse, National Institute of Mental Health, and National Institute of Allergy and Infectious Diseases Health, through the International Clinical Research Fellows Program at Vanderbilt (R24 TW007988). C. Farquhar has an NIH K24 Mentoring Award (K24 AI87399). G. John-Stewart was an Elizabeth Glaser Pediatric AIDS Foundation Scientist and has an NIH K24 Mentoring Award (K24 HD54314). J. Juno holds a CIHR Vanier Ph.D. scholarship. K. Fowke is the recipient of a CIHR New Investigator Salary Award and holds a Manitoba Research Chair from the Manitoba Health Research Council.
Written informed consent was obtained from all study participants. This study received ethical approval from the Institutional Review Boards of the University of Washington, University of Manitoba, and the Kenyatta National Hospital.
Author Disclosure Statement
No competing financial interests exist.