
Abstract
Human immunodeficiency virus (HIV) entry into susceptible cells involves the interaction between viral envelope glycoproteins with CD4 and a chemokine receptor (coreceptor), namely CCR5 and CXCR4. This interaction has been studied to enable the discovery of a new class of antiretroviral drugs that targets the envelope glycoprotein–coreceptor interaction. However, very few data exist regarding HIV-2 susceptibility to these coreceptor inhibitors. With this work we aimed to identify this susceptibility in order to assess the potential use of these molecules to treat HIV-2-infected patients and to further understand the molecular basis of HIV-2 envelope glycoprotein interactions with CCR5 and CXCR4. We found that CCR5-using HIV-2 isolates are readily inhibited by maraviroc, TAK-779, and PF-227153, while monoclonal antibody 2D7 shows only residual or no inhibitory effects. The anti-HIV-2 activity of CXCR4-targeted molecules reveals that SDF-1α/CXCL12 inhibited all HIV-2 tested except one, while mAb 12G5 inhibited the replication of only two isolates, showing residual inhibitory effects with all the other CXCR4-using viruses. A major conclusion from our results is that infection by HIV-2 primary isolates is readily blocked in vitro by maraviroc, at concentrations similar to those required for HIV-1. The susceptibility to maraviroc was independent of CD4+ T cell counts or clinical stage of the patient from which the virus was obtained. These findings indicate that maraviroc could constitute a reliable therapeutic alternative for HIV-2-infected patients, as long as they are infected with CCR5-using variants, and this may have direct implications for the clinical management of HIV-2-infected patients.
Introduction
T
In an effort to provide additional therapeutic options for HIV infection, new drugs are being developed that target viral entry into cells. 2 One of the most promising new classes of entry inhibitors includes small molecules targeting the interaction between the HIV SU glycoprotein and the chemokine receptor, CCR5. Targeting CCR5 was a logical option because CCR5, together with CXCR4, is an important coreceptor for HIV-1 entry 3,4 and also because a 32-base pair deletion in the ccr5 gene renders individuals homozygous for this deletion almost completely resistant to HIV infection. 5,6 Consequently, several CCR5-targeted antagonists have been produced and their therapeutic potential evaluated; one of them, maraviroc (MVC), 7 is already approved for the treatment of HIV-1-infected patients. 8
The well-known diversity of HIV Env glycoproteins implies that not all viral isolates interact with CD4 and coreceptors in exactly the same way. Env glycoprotein plasticity and the expected gradients of inhibition efficacy lead us to anticipate that HIV susceptibility to entry inhibitors is type, subtype, an even strain specific 7 and studies addressing this issue are obviously warranted.
Very few available data exist about human immunodeficiency virus 2 (HIV-2) susceptibility to entry inhibitors, or the influence of coreceptor inhibitors on HIV-2 evolution concerning coreceptor usage and virulence. In HIV-2 infection the diversity of coreceptor usage has been described as broader and very heterogeneous, suggesting that HIV-2 Env glycoproteins might possess an increased flexibility when compared to HIV-1. In fact, in primary HIV-2 isolates the promiscuous use of coreceptors, 9 –11 the existence of CCR5/CXCR4-independent strains, 12,13 and CD4-independent infection have been reported. 14,15
Although HIV-2 infection is endemic in West African countries such as Guinea-Bissau and Senegal, it has spread to other countries such as France and Portugal. In the latter, HIV-2 infection accounts for about 3% of reported AIDS cases. 16 In such countries, data regarding HIV-2 sensitivity to coreceptor inhibitors are crucial. It is also essential to ascertain the consequences of coreceptor inhibitors on viral dynamics and in the evolution of the HIV-2 viral population within an infected individual.
Here our goal was to analyze the in vitro susceptibility of HIV-2 to CCR5 and CXCR4 inhibitors, namely monoclonal antibodies, natural ligands, and antagonists. We tested these inhibitors using HIV-2 primary isolates with distinct phenotypes that have been obtained from patients at different clinical stages. Our data indicate that maraviroc, PF-227153 (a close analogue of PF-232798, a follow-up second-generation antagonist for maraviroc 17 ), and TAK-779 readily inhibit CCR5-using HIV-2 isolates, whereas monoclonal antibody 2D7, which interacts with the second extracellular loop of CCR5, 18 –20 inhibits only one of these isolates (HIV-2ALI), showing residual or no inhibitory effects with all the other primary isolates tested.
Materials and Methods
Cells
Peripheral blood mononuclear cells (PBMCs) from patients and donors were isolated, phytohemagglutinin-stimulated, and cultured as described. 13 PBMCs used to establish viral stocks were obtained from the same group of donors, to avoid interindividual variations in HIVxinfection susceptibility, and screened for Δ32 deletion in the ccr5 gene prior to use, as previously described. 12 A signed informed consent was obtained from all the PBMC donors, in accordance with the Portuguese National Blood Institute's Ethics Committee.
Human osteosarcoma GHOST-derived cell lines expressing coreceptors (GHOST-CD4/CCR5 and GHOST-CD4/CXCR4) were maintained in Dulbecco's modified Eagle's medium (Invitrogen, Paisley, UK) supplemented with 10% (v/v) inactivated fetal bovine serum, 50 μg/ml gentamicin, 2.5 μg/ml amphotericin B, 1 μg/ml puromycin, 100 μg/ml hygromycin, and 500 μg/ml G418 (Invitrogen, Paisley, UK).
Viruses
Primary HIV-2 isolates (HIV-2ALI, HIV-2UCFL2001, HIV-2UCFL2003, HIV-2UCFL2060, HIV-2UCFL2061, and HIV-2UCFL2070) were isolated and characterized as described. 14,21,22 HIV-1LAI 23 and HIV-1Ba-L 24 are laboratory-adapted HIV-1 strains with exclusive CXCR4 and CCR5 tropism, respectively. 25 Viral replication was assessed by reverse transcriptase activity in culture supernatants by an enzyme-linked immunosorbent assay (Lenti-RT kit, Caviditech, Uppsala, Sweden). Viral stocks were established from low-passaged supernatants of infected PBMCs and stored at −80°C.
Inhibitors and antibodies
The inhibitors TAK-779, 26 AMD3100, 27 and maraviroc 7 and the monoclonal antibodies (mAb) 2D7 20 and MAB 12G5 28 were all obtained through the National Institute of Health (NIH) AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH. The CCR5 inhibitor PF-227153 was kindly donated by Pfizer Global Research and Development-Sandwich Laboratories, Sandwich, Kent, UK. Inhibition assays also included the CXCR4 natural ligand, stromal-cell-derived factor-1, or SDF-1α/CXCL12 29,30 (R&D Systems; Minneapolis, MN).
Inhibition assays
Virus sensitivity to the different inhibitors used was performed using GHOST-CD4/CCR5 or GHOST-CD4/CXCR4 cell lines and was based on the inhibition of viral production as described. 12 Cells were seeded at 1.5×105 cells per well in 24-well plates and allowed to adhere overnight. Cells were incubated for 1 h at 37°C with different concentrations of each inhibitor plus vehicle control (the solvent used for each compound). Viruses (100 TCID50 in a final volume of 100 μl/well) were then added and incubated for 4 h in inhibitor-containing medium (plus vehicle control), in the presence of 3 μg/ml of polybrene (Sigma-Aldrich, MO). Cells were then washed with PBS to remove unadsorbed viral particles and cultured in appropriate medium (500 μl/well) containing the desired concentration of each inhibitor. Virus production was assessed every day, by RT activity in culture supernatants by an enzyme-linked immunosorbent assay (Lenti-RT kit, Caviditech, Uppsala, Sweden) until day 7 postinfection, and the peak RT activity was measured during this time period for each virus–inhibitor combination used to calculate the inhibitory activity of each compound. Infection inhibition in the presence of an inhibitor was calculated as 100× [1 – (RTInhib/RTcontrol)], the control being infection in the absence of inhibitors (vehicle only). The geometric mean 50% and 90% inhibitory concentration values (IC50 and IC90, respectively) were obtained from individual data sets fit to a sigmoidal dose–response curve by nonlinear regression using the Prism 4 program (GraphPad Software, San Diego, CA).
Results
HIV-2 susceptibility to CCR5 inhibitors
Coreceptor usage of HIV-2 isolates used was previously described 14,21,22 and summarized in Table 1. To further decipher how inhibitors targeting the CCR5 chemokine receptor affect the replication of HIV-2, we infected GHOST cell lines coexpressing CD4 and CCR5 with primary HIV-2 isolates (specified in Table 1) and HIV-1Ba-L and HIV-1LAI (as controls) in the presence of different concentrations of CCR5 inhibitors: TAK-779, maraviroc (MVC), PF-227153, and monoclonal antibody (mAb) 2D7. GHOST cell lines were used instead of PBMCs because although PBMCs represent an ex vivo (and thereby nonrecombinant) cell system, they present some disadvantages. Namely, PBMCs are constituted by a heterogeneous cell population with different levels of cellular receptors expression, which obviously will influence viral infectivity and inhibitor susceptibility. Furthermore, as shown in Table 1, the HIV-2 isolates used display distinct biotypes regarding coreceptors usage: some of them are able to interact with the CXCR4 coreceptor in addition to CCR5. A cell line such as GHOST-CD4/CCR5, expressing only the CCR5 coreceptor, will allow the precise assessment of CCR5 inhibition without interference of additional chemokine receptors present in PBMCs that could mediate HIV-2 infection. The extent of inhibition of viral replication is presented as a percentage of control (no inhibitor=0%).

Type of HIV isolate: P, primary; LA, laboratory adapted.
Clinical stage according to CDC classification.
Number of CD4+ T-lymphocytes/μl; ND, not determined.
Coreceptor usage assessed in GHOST-CD4 cells expressing CCR5 or CXCR4 coreceptors; peak RT activity was measured in culture supernatants during a 21-day period after virus inoculation: −, peak RT activity<10 pg/ml; +, peak RT activity between 10 and 100 pg/ml; ++, peak RT activity between 101 and 1000 pg/ml; +++, peak RT activity >1000 pg/ml.
R5, ability to infect GHOST cells expressing CCR5; R5X4, ability to infect GHOST cells expressing CCR5 and CXCR4; X4, ability to infect GHOST cells expressing CXCR4.
Downward arrow, increasing performance of CXCR4 coreceptor usage.
The results show that all HIV-2 isolates tested were efficiently inhibited by TAK-779, PF-227153, and MVC (Fig. 1). The compound TAK-779 was the least effective, particularly for UCFL2070 and UCFL2003 isolates (Table 2) where the IC50 was appreciable higher (18.080±0.060 and 15.799±0.367 nM, respectively) compared to ALI (3.276±0.004 nM), UCFL2001 (3.112±0.01 nM), and UCFL2061 (2.904±0.041 nM). The UCFL2060 isolate shows an intermediate susceptibility to TAK-779 with an IC50 value of 8.967±0.099 nM.
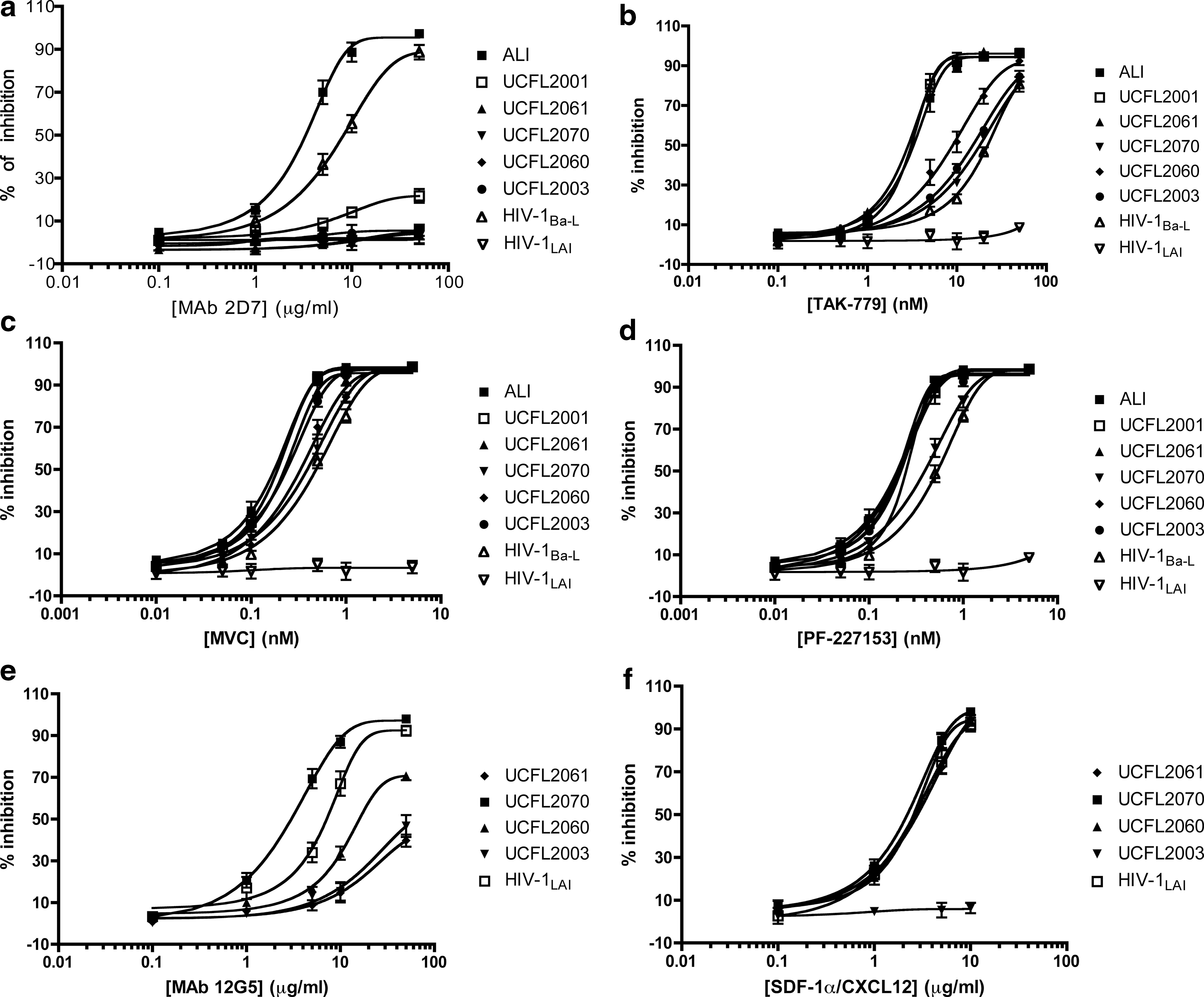
Dose–response curves for HIV-2 susceptibility to different CCR5- and CXCR4-targeted inhibitors. In these assays, HIV-2 primary isolates were normalized by RT activity levels, and equivalent amounts of each isolate were added to GHOST-CD4/CCR5
Duplicate wells were used to derive each experimental value in each experiment and the data shown are the average±standard error of the mean (SEM) of values from three independent experiments; ND, not determined. HIV-1Ba-L and HIV-1LAI strains were used as controls.
One major concern regarding the interpretation of our data was the belief that the susceptibility of HIV infection to small-molecule CCR5 inhibitors is apparently influenced by the cell type used, being smaller in cell lines engineered to express CCR5 (e.g., GHOST-CD4/CCR5) when compared to PBMCs. 31 –34 In spite of this, we did not notice significant changes in the IC50 of HIV-1Ba-L assessed in the GHOST-CD4/CCR5 cell line (Table 2) when compared to the described IC50 for the CCR5 inhibitors tested using PBMCs as target cells: TAK-779 26,35 and MVC. 7 As the potency of inhibition is conserved between the HIV-1 and HIV-2 isolates in this study, and as reported independently (and in PBMCs) for HIV-1, extrapolation suggests that the anti-HIV-2 activity for maraviroc may be clinically significant.
Noteworthy, the percent inhibition by TAK-779, PF-217153, and MVC was in general greater for HIV-2 isolates than for HIV-1Ba-L; this is particularly evident for ALI, UCFL2001, and UCFL2061 isolates (Fig. 1). Reflecting and reinforcing this notion, the values of IC50 and IC90 of TAK-779, MVC, and PF-227153 were consistently lower for HIV-2 isolates than for HIV-1Ba-L control (Table 2). However, this higher susceptibility to CCR5 inhibitors observed in HIV-2 isolates could be solely a consequence of the fact that we are comparing a laboratory-adapted strain (HIV-1Ba-L) with primary HIV-2 isolates. Extensive adaptation to in vitro T cell culture may influence coreceptor affinity and fusion process kinetics, as described for coreceptor or fusion inhibitors in HIV-1. 31,34,36,37
While TAK-779, PF-227153, and MVC show a similar capacity to inhibit HIV-2 isolates (the only exception was the UCFL2003 isolate that was much more susceptible to inhibition by MVC and PF-227153 than by TAK-779), the mAb 2D7 only inhibited the replication of HIV-2ALI, (IC50=3.347±0.017 μg/ml) in accordance to our previous data. 12 With all the other HIV-2 isolates tested, mAb 2D7 shows only residual levels of inhibition or no inhibitory effect (Fig. 1). Particularly interesting was the susceptibility behavior of the UCFL2001 isolate to mAb 2D7. This isolate has an R5 biotype as HIV-2ALI and yet it is only slightly inhibited by this monoclonal antibody specific for the CCR5 coreceptor (IC50>50 μg/ml), while remaining fully permissive to inhibition by TAK-779, PF-227153, and MVC (Fig. 1 and Table 2). Of note, different susceptibilities of HIV-2 isolates to mAb 2D7 could not be associated with disease stage since resistance to this inhibitor was found in both symptomatic and asymptomatic patients (Table 1). Also, the R5 vs. R5X4 biotype or the relative efficiencies with which CCR5 or CXCR4 coreceptors are used by HIV-2 isolates (Table 1) did not account for these different susceptibilities.
HIV-2 susceptibility to CXCR4 inhibitors
The anti-HIV-2 activity of CXCR4-targeted molecules was investigated using the GHOST-CD4/CXCR4 cell line in the presence of a range of CXCR4 natural ligand, SDF-1α/CXCL12, 29,30 and mAb 12G5 concentrations. 28 Inhibition assays performed with CXCR4-using isolates reveal that SDF-1α/CXCL12 readily inhibited all HIV-2 tested except UCFL2003 (Fig. 1f and Table 2). In contrast, mAb 12G5 (Fig. 1e and Table 2) inhibited the replication only of UCFL2070 and UCFL2060 (IC50=3.54±0.268 μg/ml and 16.859±0.792 μg/ml, respectively), showing residual inhibitory effects with all the other CXCR4-using viruses (IC50>50 μg/ml). The mAb 12G5 is able to block HIV-1 and HIV-2 infections through a specific binding to the second extracellular loop (ECL2) of CXCR4, although additional regions of this coreceptor may also influence its binding. 28,38,39 The identification of CXCR4-using primary HIV-2 isolates able to infect GHOST-CD4/CXCR4 cells, even in the presence of high concentrations of mAb 12G5, indicates that these isolates can either use CXCR4 molecules that fail to bind the mAb 12G5, probably as a result of coreceptor heterogeneity, or utilize a different region of CXCR4 coreceptor that is not blocked by this mAb. A similar mechanism was also suggested to explain the lack of antiviral activity of mAb 12G5 against some laboratory-adapted strains of HIV-1 and HIV-2. 39
The HIV inhibition by the CXCR4 natural ligand SDF-1α/CXCL12 29,30 is mediated by a different mechanism, mainly involving the internalization of the CXCR4 coreceptor. 40 Noteworthy, UCFL2003 reveals an almost complete resistance to SDF-1α/CXCL12 (Fig. 1f), a fact that together with the partial inhibition by mAb 12G5 (Fig. 1e) could suggest that this isolate may be using an unidentified coreceptor present in the GHOST-CD4/CXCR4 cell line. To exclude this possibility, we repeated the infection of GHOST-CD4/CXCR4 cells in the presence of the CXCR4-specific antagonist AMD3100 (1000 nM) and the replication was completely abrogated (data not shown). This lack of inhibition by both SDF-1α/CXCL12 and mAb 12G5, observed with the UCFL2003 isolate, was already described for an HIV-1 strain (HIV-1NL4-3) made resistant to SDF-1α/CXCL12 after serial passages in vitro. 41 The emergence of this resistant variant involves several mutations in the env gene, apparently enabling the use of a different binding site on the CXCR4 molecule. 41
Discussion
In this study we determined the susceptibility of HIV-2 primary isolates to different CCR5 and CXCR4-targeted inhibitors, including maraviroc, the first CCR5 antagonist approved for the treatment of HIV-1-infected patients. 8 Identifying the ability of these inhibitors to interfere with HIV-2 replication in vitro is important to assess the potential use of these molecules as therapeutic alternatives to treat HIV-2-infected patients, and for understanding the molecular basis of the HIV-2 interaction with CCR5 and CXCR4, the two main coreceptors for HIV infection.
The chemokine receptors inhibitors, either small molecule antagonists, natural ligands, or monoclonal antibodies, bind to the specific chemokine receptor present at the cell membrane, which acts as coreceptor together with the CD4 molecule to mediate the fusion and entry of HIV into the target cell. By doing so, they inhibit the interaction between the HIV envelope glycoprotein complex with the coreceptor, thus blocking the fusion process and interrupting the viral replication cycle. 42 In this work we show that CCR5-using HIV-2 isolates are readily inhibited by maraviroc (MVC), PF-227153, and TAK-779, whereas mAb 2D7 inhibits only one of these isolates (HIV-2ALI), showing residual or no inhibitory effects with all the other primary isolates tested.
The mAb 2D7 is a murine monoclonal antibody that shows a potent inhibitory effect on R5 and R5X4 (dual tropic) HIV-1 binding and infection. 20 This mAb recognizes a conformational epitope involving nonlinear regions of the second extracellular loop (ECL2) of CCR5. 18 –20 TAK-779 was the first nonpeptide CCR5 inhibitor 26 and its binding site has been mapped to a pocket formed between transmembrane helices 1, 2, 3, and 7 of CCR5. 43,44 Similarly, MVC a small-molecule CCR5 antagonist approved for the treatment of HIV infection, 7 appears to interact with key amino acid residues in transmembrane regions 2, 3, 5, 6, and 7 of CCR5. 45 The binding regions described for mAb 2D7, TAK-779, and MVC overlap one of the CCR5 domains, which contributes to its HIV coreceptor activity. Studies using chimera containing segments of CCR5 and synthetic peptides from extracellular regions of CCR5 have led to the general conclusion that the amino-terminal domain and the ECL2 seem to play a crucial role in CCR5-mediated entry of HIV in vitro. 20,46 –51
Our data regarding HIV-2 primary isolates inhibition by mAb 2D7 proved to be interesting. Particularly noteworthy was the unique effectiveness of HIV-2ALI inhibition by mAb 2D7 compared to other primary isolates. Knowing that 2D7 interacts with ECL2 of CCR5, this dependency on ECL2 availability suggests that this region of the CCR5 molecule is crucial for infection by this R5 strain. This kind of strain specificity of the ECL2 region has already been described for HIV-1 strains. For example, grafting of the CCR5 ECL2 onto coreceptor-inactive murine CCR5 conferred coreceptor activity for the R5 strains ADA and YU2 but not for R5X4 strain 89.6. 51 This apparent dependency may suggest that interaction with ECL2 is a requisite sine qua non for infection through the CCR5 coreceptor by R5 viruses such as ADA, YU2, and in our study, ALI, 14, 21 and it cannot be bypassed by alternative binding to other regions of the CCR5 molecule.
The dominant route to resistance to CCR5 inhibitors appears to involve the acquisition of sequence and/or conformational changes that render SU capable of interacting with CCR5 despite the presence of the inhibitor. 31,52,53 Despite several reports addressing this issue, to our knowledge, naturally occurring primary HIV isolates with intrinsic resistance to 2D7 have not been described until now. Obviously, a main question arises: why do five out of six HIV-2 primary isolates, obtained from patients completely naive to any anti-CCR5 drug, show a natural resistance to mAb 2D7, while remaining fully susceptible to other CCR5 target inhibitors, such as TAK-779 and MVC, and in experimental conditions imposing the absence of any coreceptor other thanCCR5? The generation after several passages in vitro of escape mutants with significant resistance to mAb 2D7 was described for HIV-1JR-CSF. 54 Although the genetic determinants and the mechanisms of resistance were not fully identified, it appears that the ability to evade the block to entry, imposed by the presence of mAb 2D7, could, at least in some circumstances, be mediated by a shift in the CCR5 domain that is used for viral entry into cells. This escape mechanism was further referred in vitro for mAb3952 directed (as mAb 2D7) to the ECL2 of CCR5, 55 and for small-molecule inhibitors, 32,52,56 and apparently involves the switch of the preferential binding region from ECL2 (wild-type virus) to the amino-terminal region (resistant virus).
During HIV interaction with cellular receptors, several conformational changes in both viral envelope glycoproteins and chemokine receptor are needed for the fusion to take place. In particular, the SU glycoprotein interaction with coreceptor seems to be a multistep process involving the sequential interaction with the amino-terminal region followed by the ECLs (namely ECL2). Thus, it is conceivable that in the HIV-2ALI isolate this sequential interaction is abrogated by the hindrance caused by the presence of mAb 2D7 in the ECL2 while all the other HIV-2 isolates have an SU conformation that apparently allow them to predominantly interact with other CCR5 regions (such as the amino-terminal) or alternatively provide the capacity to maintain a significant interaction with ECL2 even in the presence of mAb 2D7.
Noteworthy, the isolation of these 2D7-resistant variants could hardly be the result of an in vitro selection as they were isolated and propagated in the complete absence of any CCR5 inhibitor, including mAb 2D7. Furthermore, the 2D7-resistant HIV-2 isolates do not show any loss of fitness as assessed by a replicative kinetics rate compared to the 2D7-susceptible isolate, HIV-2 ALI (data not shown), 12,21 suggesting that the structural changes required to overcome the presence of mAb 2D7 did not compromise the dynamics of the viral fusion process either because they do not involve critical regions of the envelope glycoproteins or because they were compensated by concomitant changes in other regions of these glycoproteins. The flexibility of the CCR5 conformation should also account for this resistance, and the effect of inhibitor interaction is crucial for inducing different conformations in the CCR5 molecule, some of which will be more critical for envelope glycoprotein binding than others. 44 In addition, it will be important to determine if this natural resistance to mAb 2D7 is related to a peculiar envelope glycoprotein flexibility apparently common in HIV-2 isolates, or is the result of some in vivo pressure driving the selection of viral variants able to interact preferentially with other CCR5 regions other than ECL2.
Although the exact mechanisms are not yet fully understood, it has been suggested that antibodies and antagonists bound to CCR5 may affect the structure of this chemokine receptor and that the binding of these inhibitors of CCR5 may disturb the conformational changes required during the fusion process. 57 The unchanged susceptibility of HIV-2 isolates to small-molecule inhibitors TAK-779, PF-227153, and MVC, despite the resistance to mAb 2D7, suggests that these inhibitors may more efficiently prevent binding and fusion events during viral interaction with target cells than the mAb 2D7. This could be related to differences in the binding sites and mechanisms of action of TAK-779, PF-227153, and MVC compared to mAb 2D7. Further studies to address the exact mechanism and the molecular features underlying this phenotype are warranted.
Despite the limited number of HIV-2 strains included in this study, we may further conclude that infection of susceptible cells by HIV-2 primary isolates is readily blocked by maraviroc at concentrations similar to those required for HIV-1. The susceptibility to maraviroc was independent of CD4+ T cell counts or clinical stage of the patient. This is the first report demonstrating the suitability of maraviroc as an antagonist for primary HIV-2 isolates in vitro. These findings may have direct implications for the clinical management of HIV-2-infected patients and reinforce the recently described clinical efficacy of maraviroc as part of salvage therapy in HIV-2-infected patients. 58
Footnotes
Acknowledgments
The following reagent was obtained through the AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH: CXCR4 Monoclonal Antibody (mAb) 12G5 from Dr. James Hoxie; HIV-1LAI from Dr. Jean-Marie Bechet and Dr. Luc Montagnier; HIV-1Ba-L from Dr. Suzanne Gartner, Dr. Mikulas Popovic, and Dr. Robert Gallo; maraviroc (Cat. #11580); TAK-779; AMD3100 and mAb 2D7. The CCR5 inhibitor PF-227153 was kindly donated by Pfizer Global Research and Development-Sandwich Laboratories, Sandwich, Kent, UK. The GHOST indicator cell lines expressing CD4 and chemokine receptors CCR5 and CXCR4 (repository numbers: ARP078 and ARP079, respectively) were obtained from the Programme EVA Centre for AIDS Reagents, NIBSC, UK, supported by the EC FP6/7 Europrise Network of Excellence, AVIP and NGIN consortia, and the Bill and Melinda Gates GHRC-CAVD Project and was donated by Dr. Littman and Dr. V KewalRamni (courtesy of NIH AIDS Research and reference Reagent Programme). This work was supported by grants from Fundação da Ciência e Tecnologia (PPCDT/SAU-IMI/55726/2004); Coordenação Nacional para a Infecção VIH/SIDA—Alto Comissariado da Saúde (CNIVS 28-1.7.3/2004); Associação Nacional de Farmácias; Merck Sharp & Dohme Portugal; and Gilead Sciences Portugal.
Author Disclosure Statement
No competing financial interests exist.