
Abstract
The foreskin contains a subset of dendritic cells, macrophages, and CD4+ and CD8+ T cells that may be targets for initial HIV infection in female-to-male sexual transmission of HIV-1. We present analyses comparing HIV-1 sequences isolated from foreskin DNA and serum RNA in 12 heterosexual men enrolled in an adult male circumcision trial performed in Rakai, Uganda. Phylogenetic analysis demonstrated three topologies: (1) little divergence between foreskin and serum, (2) multiple genetic bottlenecks occurring in both foreskin and serum, and (3) complete separation of foreskin and serum populations. The latter tree topology provided evidence that foreskin may serve as a reservoir for distinct HIV-1 strains. Distance and recombination analysis also demonstrated that viral genotypes in the foreskin might segregate independently from the circulating pool of viruses.
H
Established HIV-1 infection is characterized by a large degree of viral genetic heterogeneity due to the high mutational error rate of the virus and the varied host immunologic pressures in different anatomic or cellular reservoirs and compartments. Viral compartments are cell types or tissues in which there is a restriction of virus flow to the primary replicating viral population, while viral reservoirs are cell types or tissues in which replication-competent viruses accumulate and persist with more stable kinetic properties than in the primary pool of actively replicating virus. 3 Reservoir sequence populations often have higher levels of genetic diversity than circulating replicating viruses obtained at the same time point due to the low level generation of new viral species and the persistence of historical viral species. 4 Viral compartments and reservoirs have the potential to occasionally reseed the periphery with genetically distinct viral variants, thereby increasing the chance for immune escape, development of drug-resistant mutations, and reduced ability to control the infection. 3 In this study, our goal was to employ a sequence analysis approach to determine if HIV-1 viral variants found in the male foreskin differ from those in circulation.
The study population consisted of HIV-1-positive control arm men enrolled into the male circumcision trial conducted in Rakai, Uganda, in 2003–2007. Control men were offered and accepted circumcision after 24 months of follow-up. Enrollment and study details have been previously described. 5 Institutional Review Board approvals were obtained from the Uganda Virus Research Institute's Science and Ethics Committee, Uganda National Council for Science and Technology, Western IRB, and Johns Hopkins University, Bloomberg School of Public Health. All participants provided written informed consent prior to study enrollment. Prior to surgery, a medical assessment was performed, venous blood was drawn, and serum extracted and stored at −80°C. At surgery, foreskins were removed and preserved in 70% ethanol at −80°C. In this study, 12 men with moderate to high plasma viremia (>10,000 copies/ml) were evaluated.
Serum samples were extracted and amplified as previously described 6 with forward primer relative to HXB2 positions 5958–5977 (TAG GCA TYT CCT ATG GCA GG) and reverse primer relative to positions 7792–7822 (AGT GCT TCC TGC TGC TCC CAA GAA CCC AAG). Subsequently, nested primers were used relative to HXB2 positions 6197–6216 (ATW AGN GAA AGA GCA GAA GA) and reverse primer relative to positions 7667–7647 (CAC TTC TCC AAT TGT CCC TCA) using identical conditions. Total DNA was extracted from foreskin samples using the QIAGEN QIAamp DNA mini kit. PCR amplification was performed for foreskin as with the serum without using a reverse transcription step. The ∼1500-bp PCR products were gel purified and cloned (Invitrogen Zero Blunt TOPO PCR kit). The transformed cells were then incubated on kanamycin selective media plates at 30°C for 48 h with controls. Bacterial culture plates were submitted for DNA nucleotide sequencing (Genewiz Inc., South Plainfield, NJ). Sequences from all individuals were aligned with the Clustal W algorithm and were further optimized by hand. An all-inclusive neighbor-joining phylogenetic tree was constructed from gap-stripped sequences to verify that each subject's sequence population segregated independently. Based on the NCBI genotyping tool, all sequences were subtype D with the exception of subject N17873, who was infected with HIV-1 subtype A1 and patient N15661, who was infected with HIV recombinant form A1/D. The sequences were deposited in GenBank (JF932509–JF933732).
Maximum-likelihood (ML) trees were inferred using the program PhyML and the best fitting nucleotide substitution model determined by a hierarchical likelihood ratio test using the HKY85 substitution and gamma rate models. Statistical support for internal branches of each tree was obtained by bootstrapping (500 replicates). ML trees were mid-point rooted and color-coded using Figtree software (
Maximum-likelihood phylogenies demonstrated three patterns of viral segregation among subjects' sequences: (1) One subject displayed little significant separation at any point in the tree between serum and foreskin samples or significant branches within the overall sequence population (N18674) (Fig. 1A). This subject was infected less than 6 months prior to circumcision and the phylogeny suggests a close association between viruses in both tissues. (2) Sequence populations from eight subjects had significant clades containing either foreskin or serum sequences that were distributed throughout the tree and in these clades frequent segregation points were observed (L14622, N21656, N15661, M13181, M14720, M13806, M15342, and N21096) (Fig. 1B). (3) Sequence populations from three subjects exhibited clear separation of serum and foreskin sequences with little segregation in serum samples and multiple distinct strains in the foreskin sequence populations (M16827, M17505, and N17873) (Fig. 1C). The single subject infected with HIV-1 subtype A exhibited this third pattern of viral segregation.
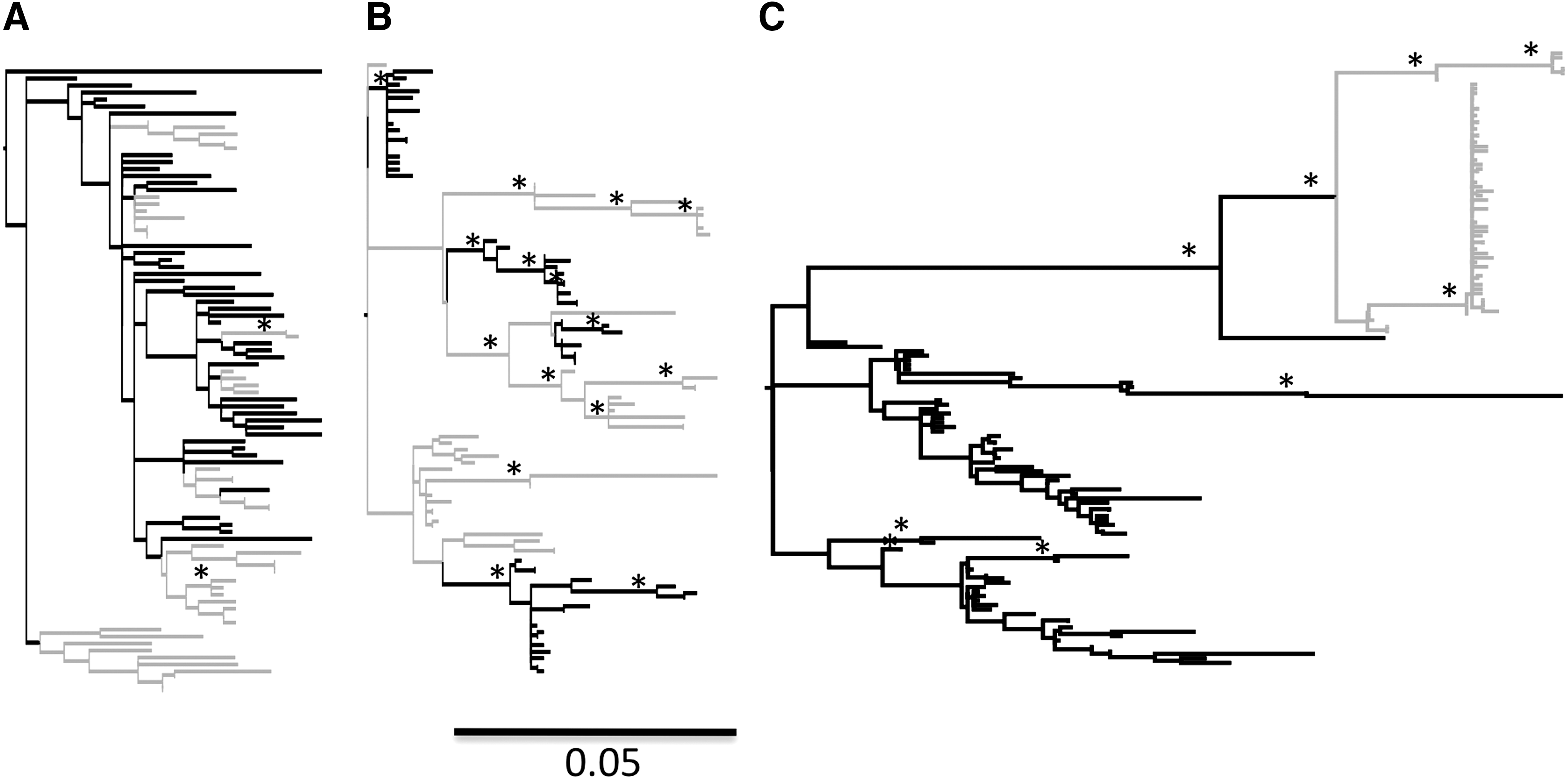
Maximum-likelihood phylogenies for patient foreskin and serum sequence populations. Maximum-likelihood midpoint-rooted phylogenies of HIV gp120 isolated from serum (gray) and foreskin (black) samples for three patterns are shown. Each panel represents an example of an observed pattern of viral segregation in a single patient in each of the following groups:
Five subjects showed no significant differences between serum and foreskin distance calculations (N18674, N21096, M13181, M15345, and N15661). Seven subjects' sequence populations showed significantly higher distances between serum and foreskin tissues (M13806, L14622, M14720, N21656, N17873, M17505, and M16827). As expected, a large distance between foreskin and serum sequences was calculated for each of the three subjects with highly segregated serum and foreskin sequence populations. dN/dS ratios showed evidence for selective pressure acting on foreskin sequence populations from two subjects (dN/dS>1.0) (N18674, M17505). All but two foreskin samples showed a higher dN/dS ratio than serum samples, although in most cases the difference was not significant (Table 1).
Ratio of nonsynonymous substitutions per site to synonymous substitutions per site.
Within group percent diversity±standard error.
Between group diversity±standard error.
Recombination analysis revealed up to 59% of the isolates in foreskin and 44% of the isolates in serum were intrasubject recombinant viruses (Table 2). Again, the three sequence serum populations corresponding to sequence populations with the phylogenetic topology shown in Fig. 1C displayed little to no signal for recombination. Overall, 26 recombinant breakpoints were identified by GARD within foreskin samples as compared to 12 in serum sequence populations. Figure 2 demonstrates a single example of recombination occurring between serum and foreskin. In Fig. 2A, a phylogenetic network is shown for sequences from patient N18674 that contains four serum and six foreskin sequences. The Phi test for recombination was significant at p=0.005. Removal of one of the serum sequences resulted in the phylogenetic network shown in Fig. 2B and an insignificant Phi test of p=0.1. Bootscanning of these sequences showed that the query sequence, which was derived from serum, shared a similar segment in V1-V2 with a serum sample and the V4-V5 region with a foreskin sequence. This patient was the most recently infected in our study set with infection occurring less than 6 months prior to circumcision. In sequence data sets corresponding to phylogenetic trees in Fig. 1A and B, recombination events between serum and foreskin were identified in all populations except M15661. Recombination events between serum and foreskin samples were not detected using the methods in Splitstree or GARD in the sequences corresponding to phylogenetic trees displayed in Fig. 1C; however, in one of these cases (N17873) we found a small segment of sequence within the sequence alignment that appears to be exchanged in a hotspot for recombination: the highly glycosylated V4 domain. In Fig. 2D this potentially exchanged sequence segment between serum and prepuce is shown. Only one foreskin sequence contained the motif NSTWDKDG, whereas all serum sequences contained this motif.
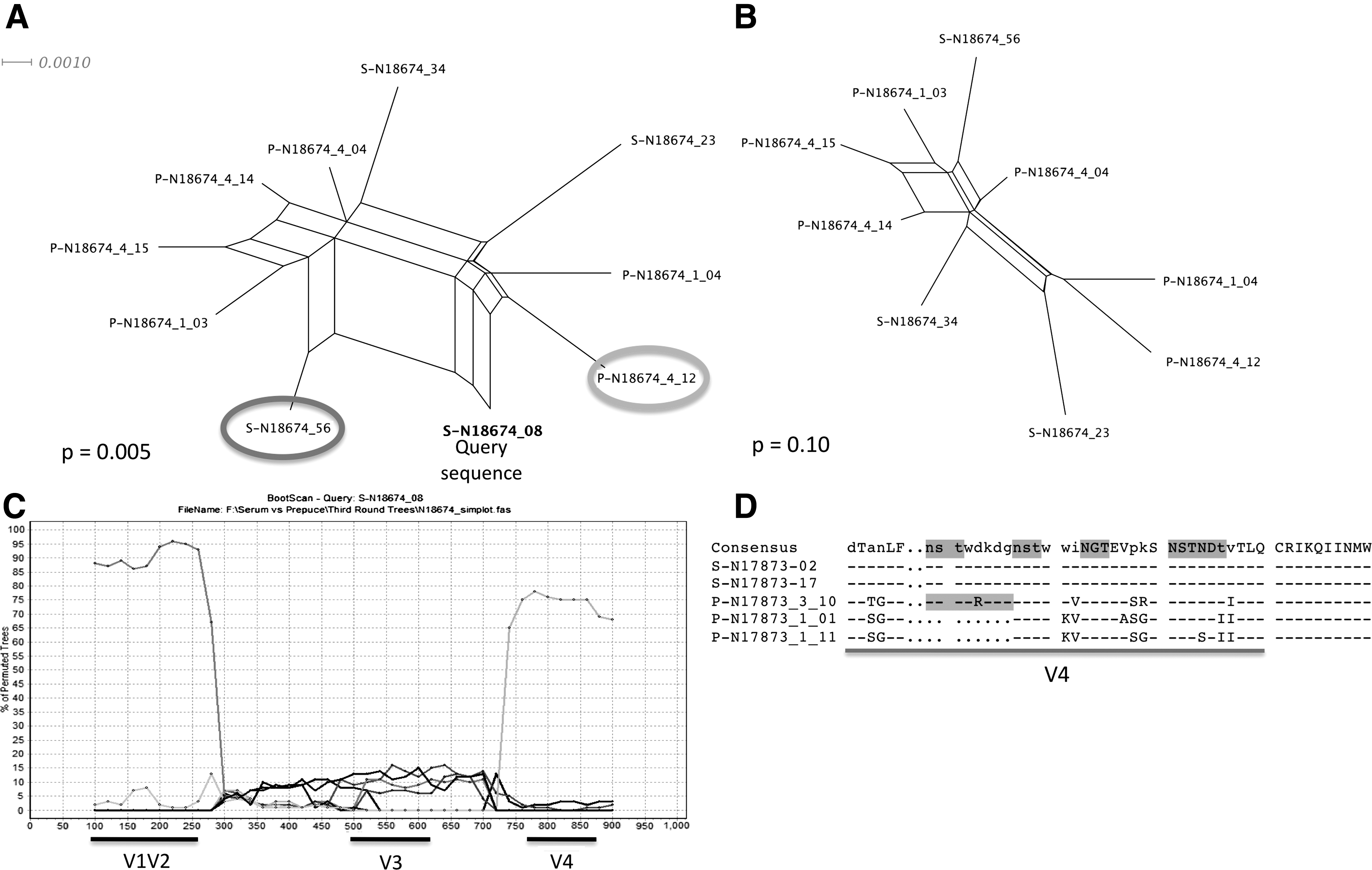
Recombination between serum and foreskin.
Percent recombinants in foreskin and serum using SplitsTree.
Location of significant breakpoints in foreskin and serum using the Genetic Algorithm Recombination Detection (GARD).
Tropism prediction analysis showed that some subjects may harbor a majority of X4 viruses (N15661, M15343, and M17505). In the subject most recently infected (N18674), potential X4 variants appeared almost equally dispersed in the phylogenetic tree between both foreskin and serum sequences. There was no relationship between tropism prediction and viral load.
The three observed segregation patterns between foreskin and serum viruses in the 12 subjects (Fig. 1) demonstrate that foreskin is not a viral compartment; however, in some cases HIV-infected cells in the foreskin may act as a reservoir for HIV. This idea is supported by analysis of the three sequence populations associated with the phylogenetic topology in Fig. 1C and corresponds to the distance and recombination calculations in the last three sequence populations listed in Tables 1 and 2. The foreskin sequences in these three subjects demonstrate the pattern of a viral reservoir as described by Nickle et al., 4 where increased sequence diversity is observed in a reservoir tissue or cell type when compared to nonreservoir sequence populations due to the presence of both recent and historical viral sequences. At the same time, the reduced degree of viral heterogeneity in the serum sequences in these three patients may suggest a highly stable circulating virus population under reduced immunological pressure.
Recombination is a major evolutionary force affecting viral genetic variation within an HIV-1-infected individual and is predicted to occur at the same order of magnitude as point mutational change 9 ; therefore, it was not surprising to find a large number of intrasubject recombinants. However, it was interesting that 7 of the 12 patients displayed an increased number of recombinants in integrated DNA within foreskin when compared to replicating RNA in the serum. Higher numbers of recombinant sequences have previously been reported in macrophage-associated brain tissues in subjects with dementia 7 ; therefore, this finding may be due to the infection of different cell types in foreskin than those infected in serum. Alternatively, the finding could be due to the persistence of diverse viral species in a tissue reservoir. The presence of recombinant sequences containing sequence fragments from both serum and foreskin in most of the cases analyzed indicates mixing or migration of viruses to and from each reservoir is possible.
HIV-1 evolves primarily in CD4+ T cells and macrophages, although infection of other cell types has been reported. 10 While the outer foreskin is similar to the epidermis of other regions of the penis, the inner foreskin is a distinct mucocutaneous tissue that is probably less keratinized and consists of stratified squamous epithelial cells interspersed with LCs. 11 LCs are antigen-presenting dendritic cells of monocyte origin that reside in the epidermis. Beneath this epidermal layer is the submucosa, which contains both tissue macrophages and T cells. LCs in the foreskin epithelium exist at a 12-fold higher density than macrophages or T cells and at a 3-fold higher density in the submucosa, thus suggesting that LCs may be the first cells that come into contact with HIV. 12 Unlike T cells, HIV-infected macrophages resist apoptosis once infected. 13 Within 2 days after sexual transmission, HIV-1 can be found in fluid draining from iliac lymph nodes and a dynamic infection of the body is in progress. 14 Recent studies have used the Trofile biological assay to evaluate tropism usage in subtype D virus populations and found that, different from subtype B viruses, CCR5, CXCR4, or dual tropic viruses are equally transmittable. 15 In our attempt to predict coreceptor usage, we also found several subjects who appeared to harbor a primarily CXCR4 viral genotype in both foreskin DNA and serum RNA. This may be true; however, only biological tropism testing would be able to confirm this finding.
The reduced rate of heterosexual transmission of HIV-1 to circumcised men demonstrates that the foreskin is vulnerable to infection by the virus. In the current study our genetic analyses suggest that in some cases HIV-1 migrates between the foreskin and the primary replicating viral pool in the blood. Additionally, viruses in each tissue may segregate from one another once an established infection is in place. These findings have implications for the control of viral populations during antiretroviral treatment and vaccine studies.
Footnotes
Acknowledgments
This work was supported by the Bill and Melinda Gates Foundation Grants 22006.02 and 22006.03, the Fogarty International Center Grant 3D43TW000010-21S2, and the Division of Intramural Research, National Institute of Allergy and Infectious Diseases, National Institutes of Health.
Author Disclosure Statement
No competing financial interests exist.