
Abstract
Rho GTPases are able to influence the replication of human immunodeficiency virus type 1 (HIV-1). However, little is known about the regulation of HIV-1 replication by guanine nucleotide dissociation inhibitors (GDIs), one of the three major regulators of the Rho GTPase activation cycle. From a T cell-based cDNA library screening, ARHGDIB/RhoGDIβ, a hematopoietic lineage-specific GDI family protein, was identified as a negative regulator of HIV-1 replication. Up-regulation of ARHGDIB attenuated the replication of HIV-1 in multiple T cell lines. The results showed that (1) a significant portion of RhoA and Rac1, but not Cdc42, exists in the GTP-bound active form under steady-state conditions, (2) ectopic ARHGDIB expression reduced the F-actin content and the active forms of both RhoA and Rac1, and (3) HIV-1 infection was attenuated by either ectopic expression of ARHGDIB or inhibition of the RhoA signal cascade at the HIV-1 Env-dependent early phase of the viral life cycle. This is in good agreement with the previous finding that RhoA and Rac1 promote HIV-1 entry by increasing the efficiency of receptor clustering and virus-cell membrane fusion. In conclusion, the ARHGDIB is a lymphoid-specific intrinsic negative regulator of HIV-1 replication that acts by simultaneously inhibiting RhoA and Rac1 functions.
Introduction
T
Members of the Rho GTPase family, including RhoA, Rac1, and Cdc42, have been reported to regulate the replication of HIV-1 at various stages of the viral life cycle, including viral entry, transcription, and viral release. 1 –7 Although these proteins are widely expressed, genome-wide screens for proteins that regulate HIV-1 replication using siRNA or shRNA libraries have failed to identify Rho GTPases. 8 –10 This may suggest that Rho GTPases are not potent regulators of HIV-1 replication and are therefore difficult to detect unless the viral replication assay is employed, since multiple replication cycles augment the biological effects of Rho GTPases.
There are three major regulators of the activation cycle of Rho GTPases: guanine nucleotide exchange factors (GEFs), GTPase-activating proteins (GAPs), and guanine nucleotide dissociation inhibitors (GDIs or ARHGDIs). RhoGEFs promote the exchange of GDP for GTP, RhoGAPs accelerate GTP hydrolysis, and ARHGDIs stabilize the GDI-bound form of Rho GTPases and also mask the lipid moiety of Rho GTPases, thereby sequestering Rho GTPases at the plasma membrane. 11 –14 Some RhoGAPs and RhoGEFs have been known to regulate HIV-1 replication. 6,9,10,15 –18 Positive regulators of Rho GTPases, such as RhoGEFs, were identified in the genome-wide screens, although these studies yielded varying results. In this sense, the upstream regulators of Rho GTPases may more potently influence HIV-1 replication than Rho GTPases themselves when expression levels are dysregulated. ARHGDIs have yet to be identified as regulators of HIV-1 replication.
Previously, we established a genetic screening system using a T cell-derived cDNA library to isolate cellular factors that render cells resistant to HIV-1 replication. 19,20 This system is unique in that the screen is based on MT-4 cells, a human CD4-positive T cell line, which were exposed to replication-competent HIV-1. Using this system, the carboxy-terminal domains of Brd4 and SEC14L1a were found to be negative regulators of HIV-1 replication. 19,20 Importantly, these factors were not found in previous genetic screens, suggesting that our system complements other genome-wide analyses. In this study, we describe a lymphoid-specific RhoGDI that negatively regulates HIV-1 replication through attenuation of both RhoA and Rac1 functions.
Materials and Methods
Cells
Cells were maintained in RPMI 1640 medium (Sigma, St. Louis, MO) supplemented with 10% fetal bovine serum (FBS; Japan Bioserum, Tokyo, Japan or Thermo Fisher Scientific Inc., Waltham, MA), 50–100 U/ml penicillin, and 50–100 μg/ml streptomycin (Invitrogen, Tokyo, Japan), and then incubated at 37°C in a humidified 5% CO2 atmosphere. The selection of cDNA library-transduced cells was described previously. 20 To select puromycin-resistant cells after infection with pQc- or pSM2c-based murine leukemia virus (MLV) vector, 1 μg/ml puromycin (Sigma) was added to the culture medium.
Plasmids
The plasmid vectors pCMMP, pMDgag-pol, pSV-tat, phRL/CMV, and pVSV-G were described previously. 21 The shRNA expression vectors pSM2c and pSM2/ARHGDIB (RHS1764-9680880) were purchased from Thermo Fisher Scientific (Open Biosystems Products, Huntsville, AL). The pQcXIP was obtained from Clontech (BD Biosciences Clontech, Palo Alto, CA). pGEX-Rhotekin-RBD and pGEX-4T-PAK2-RBD were kindly provided by S. Narumiya, Kyoto University. 3,22 To construct pLTR-hRL, the HIV-1HXB2 long terminal repeat (LTR) was amplified by PCR using the following primers: 5′-GGA TCC TGG AAG GGC TAA TTC ACT CC-3′ and 5′-GCT AGC TGC AGC TGC TAG AGA TTT TCC ACA CTG-3′. The BglII-NheI fragment of the PCR product was cloned into the corresponding restriction sites of phRL/CMV, generating pLTR-hRL. The LTR-Luc plasmid was described previously. 23 To construct pCMV-Tat-FLAG, HIV-1NL4-3 tat was amplified from cDNA prepared from HIV-1-infected MT-4 cells by RT-PCR using the following primers: 5′-ATG GAG CCA GTA GAT CCT AGA CTA GAG CCC T-3′ and 5′-TTC CTT CGG GCC TGT CGG GT-3′. The PCR product was cloned into the EcoRV sites of pcDNA3.1 Zeo(+) bearing FLAG-tags at the NotI-XhoI site (Invitrogen). The pCMV-Luc plasmid was a generous gift from Dr. Hijikata (Kyoto University).
Flow cytometry
Cells were incubated with anti-CD4, anti-CD8, or anti-CXCR4 monoclonal antibodies conjugated to R-phycoerythrin (PE; BD Pharmingen, San Diego, CA) for 30 min at 4°C. Cells were washed once with phosphate-buffered saline (PBS) supplemented with 1% FBS and analyzed by FACS Calibur (Beckton Dickinson, San Jose, CA).
Phalloidin staining of F-actin
Phalloidin staining of F-actin was performed as described previously. 24 In brief, cells were fixed, permeabilized by cytofix/cytoperm (BD Bioscience) for 20 min on ice, washed, and stained with Alexa Fluor 647-phalloidin (Invitrogen) for 30 min at 4°C. Samples were kept on ice until analysis by FACS Calibur or Canto II (Beckton Dickinson, San Jose, CA). The flow cytometric data were analyzed using FlowJo version 9.3 (Tree-star Inc., Ashland, OR).
Viruses
The retroviral vector pCMMP, carrying the MT-4 cDNA library and a green fluorescent protein (GFP) expression cassette, was obtained from Takara (Takara, Otsu, Japan). Full-length ARHGDIB was cloned from a lymph node cDNA library (Takara) by RT-PCR using the primers 5′-GCA CCG GTC TCG AGC CAC CAT GAC TGA AAA AGC CCC AGA GC-3′ and 5′-CCA ATT GGA TCC TCA TTC TGT CCA CTC CTT CTT AAT CG-3′, and cloned into the pCMMP vector. The MLV vectors were produced by tripartite transfection of pMDgag-pol, pVSV-G, and either pCMMP, pQc, or pSM2c retroviral vector. Cells were infected with the MLV vectors as described previously. 21 The production of replication-competent HIV-1HXB2 and the measurement of replication kinetics were performed as described previously. 21
Western blotting
Western blotting was performed according to techniques described previously.
25
The following monoclonal antibodies were used: anti-RhoA (C-11; Santa Cruz Biotechnology, Santa Cruz, CA), anti-Rac1 (23A8; Millipore, Japan), anti-Cdc42 (B-8; Santa Cruz Biotechnology), anti-ARHGDIB (A01; Abnoba, Taiwan), anti-p24CA (183-H12-5C; NIH AIDS Research and Reference Reagent Program), anti-actin (clone C4; Millipore), and anti-tubulin (DM1A; Sigma). Densitometric analysis was performed using ImageJ ver. 1.43 software (obtained from
Active Rho GTPase capture assay
We adopted a protocol described previously. 3,22 In brief, the glutathione S-transferase (GST) fused to the Rho GTPase binding domain (RBD) of Rhotekin and GST fused to the RBD of PAK2 were expressed in E. coli and purified by incubating the cell lysates with glutathione-Sepharose 4B beads (GE Healthcare Bio-Sciences, Piscataway, NJ) for 3 h at 4°C. The beads were washed three times with ice-cold Rho buffer (25 mM HEPES, pH 7.5, 150 mM NaCl, 10 mM MgCl2, 1 mM EDTA, 10% Glycerol). Finally, the beads were washed with ice-cold Rho buffer supplemented with 1% NP-40 and protease inhibitor cocktail tablets (Complete, Roche Diagnostics GmbH, Mannheim, Germany). Cells were incubated in ice-cold Rho buffer for 15 min on ice, and the cell lysates were clarified by centrifugation at 18,000×g at 4°C for 20 min. A fraction of the cleared cell lysates was incubated at 37°C for 30 min as a negative control or coincubated with GTPγS (0.1 mM, Sigma) for 30 min at 30°C as a positive control. These preparations were incubated with the above beads at 4°C for 1 h. The beads were washed three times with Rho buffer containing 1% NP-40 and subjected to SDS-PAGE followed by Western blotting. The bead-bound GTPases were detected using a monoclonal antibody against RhoA, Rac1, or Cdc42, as appropriate.
Single-round infection assay
Single-round virus infection and luciferase assays were performed as described previously. 21,26 In brief, cells were exposed to viral preparations containing 1–10 ng of p24CA. Luciferase activities were measured at 2–3 days postinfection with the Picagene luciferase assay kit (Toyo Ink, Tokyo, Japan) or Steady-Glo kit (Promega, Tokyo, Japan) according to the manufacturer's protocols. For the inhibition of Rho-kinase (ROCK), cells were pretreated with 12.5 μM Y27632 (Nakalai tesque, Kyoto, Japan) for 1 h and incubated with the viral preparation in the presence of 12.5 μM Y27632 for 4 h. Cells were washed with tissue culture medium and cultivated for 2 days. Light emission was detected with a 1420 ARVOSX multilabel counter (Perkin Elmer, Wellesley, MA) or a Veritas luminometer (Promega).
Single-round production assay
Five million PM1/control or PM1/ARHGDIB cells were resuspended in 250 μl of STBS (25 mM Tris-Cl, pH 7.4, 137 mM NaCl, 5 mM KCl, 0.6 mM Na2HPO4, 0.7 mM CaCl2, and 0.5 mM MgCl2), including 10 μg of pHXB2 proviral plasmid DNA. Cells were mixed with 250 μl of STBS containing 1 mg/ml of DEAE-Dextran and incubated for 30 min at room temperature (RT). The cells were then incubated with STBS containing 10% DMSO for 2 min at RT and washed with 1 ml HBSS (Invitrogen). Transfected cells were cultured in RPMI medium containing 10% FBS and 1 μM efavirenz. After 2 days in culture, viruses in the culture supernatant were pelleted by ultracentrifugation on a 20% sucrose cushion. Cells and viruses were lysed with radioimmunoprecipitation assay (RIPA) lysis buffer (0.05 M Tris-HCl, 0.15 M NaCl, 1% Triton X-100, 0.1% sodium dodecyl sulfate, and 1% sodium deoxycholate) and electrophoresed in a 10% polyacrylamide gel for SDS–PAGE. Proteins were then transferred to a PVDF membrane, and p24CA was detected by immunoblot analysis using an anti-p24CA antibody.
Reporter expression assay
The luciferase-expressing reporter plasmids were transfected into PM1 cells according to the DEAE-Dextran method as described above. For transfection, either 1 μg of pCMV-Luc, 10 μg of pLTR-Luc, 0.5 μg of phRL/CMV, or 1 μg of pLTR-hRL was used. The pLTR-Luc and pLTR-hRL were cotransfected with 5 μg of pCMV-tat-FLAG and 1 μg of pSVtat, respectively. Firefly luciferase activity was measured as described above. Renilla luciferase activity was also measured as described above, except that the Renilla Luciferase Assay System (Promega) was used in place of the Steady-Glo kit. The data were analyzed using a two-tailed Student's t-test.
Results
Identification of RhoGDIβ/ARHGDIB as a negative regulator of HIV-1 replication
A pool of MT-4 cells constitutively expressing a cDNA library transduced with an MLV-based retroviral vector was used to screen for possible regulators of HIV-1 replication. The MLV vector carried an expression cassette for GFP and inserts from a cDNA library derived from MT-4 cells (Fig. 1A). The cDNA-transduced cells were enriched with a cell sorter using GFP as a marker. These cells were then infected with CXCR4-using (X4) HIV-1HXB2. MT-4 cells have been shown to support efficient HIV-1 production and rapidly undergo cell death after infection. The surviving MT-4 cells were propagated and genomic DNA was isolated to identify the inserted cDNA, as described previously. 19 These genes were considered potential negative regulators of HIV-1 replication. A cDNA clone encoding ARHGDIB (RhoGDIβ/LyGDI/D4GDI; gene ID 397) was recovered from the MT-4 cDNA library (1/94 independent clones, 1.1%).
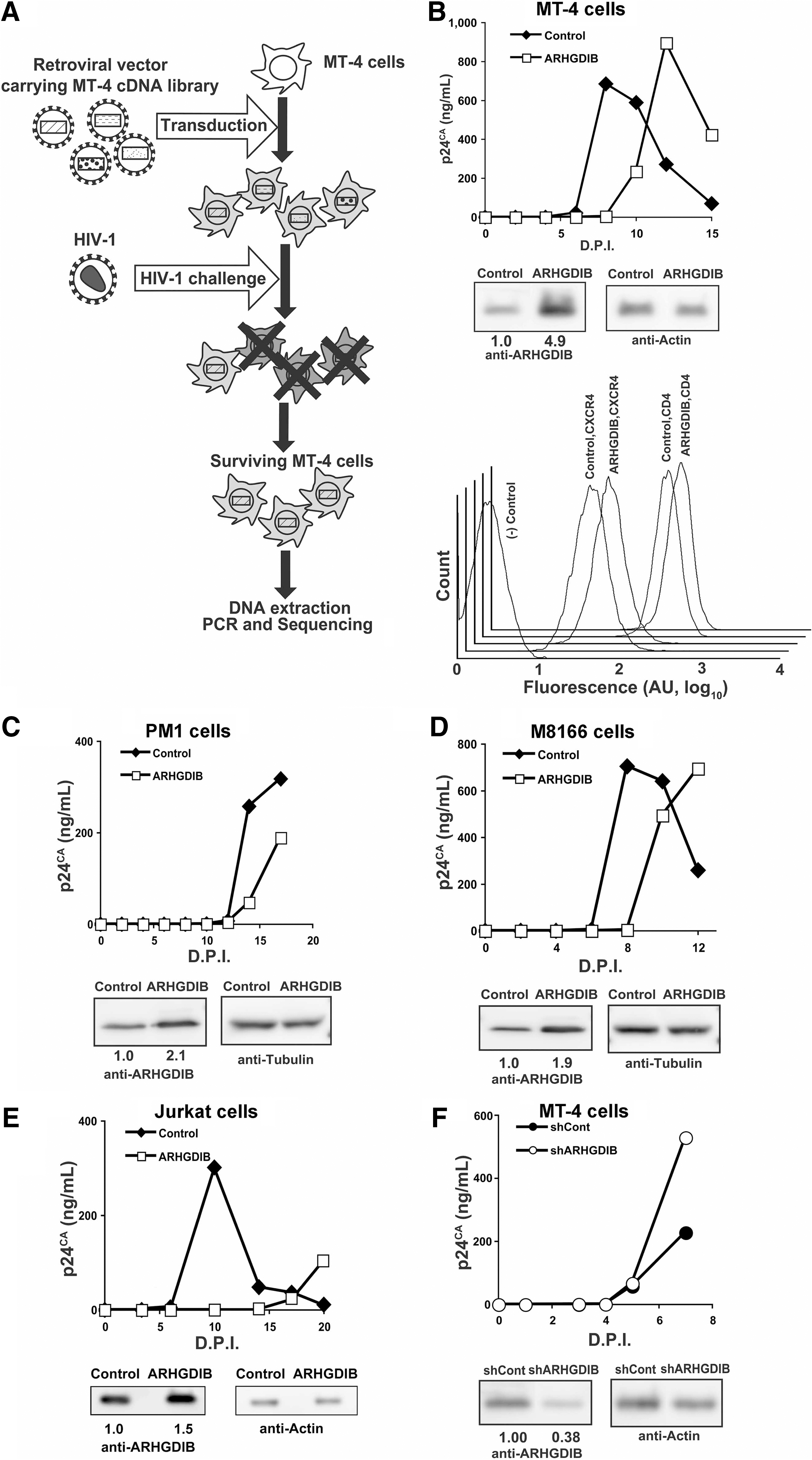
Isolation and characterization of ARHGDIB as a negative regulator of HIV-1 replication.
Full-length ARHGDIB cDNA was cloned into the MLV vector pQcXIP, and a T cell line expressing ARHGDIB at levels higher than endogenous levels was established. To verify the HIV-1-resistant phenotype of ARHGDIB, the rate of HIV-1 replication was assessed in MT-4 cells subjected to ectopic ARHGDIB expression. In these cells, ARHGDIB levels were increased by approximately 4.9-fold compared with control cells, while the cell surface expression of CD4 and CXCR4 was not significantly affected in MT-4 cells (Fig. 1B). Even the modest up-regulation of ARHGDIB delayed HIV-1 replication kinetics (Fig. 1B). Suppression of HIV-1 replication was independently reproduced in MT-4 cells, even when a different retroviral vector, pCMMP, was used to transduce ARHGDIB (data not shown). In addition, neither the rate of cell proliferation nor cell morphology was affected by stable ectopic expression of ARHGDIB over at least 6 months in culture (data not shown). Delayed HIV-1 replication in cells ectopically overexpressing ARHGDIB was also observed in PM1 (Fig. 1C), M8166 (Fig. 1D), and Jurkat cells (Fig. 1E), in which ARHGDIB levels were increased by 2.1-, 1.9-, and 1.5-fold, respectively, compared with control levels. Similar data were also obtained in SUP-T1 cells (data not shown). The delayed HIV-1 replication in cells ectopically expressing ARHGDIB was reproduced in 10 independent experiments (p<0.01, two-sided binomial test), strongly suggesting that ARHGDIB attenuates the replication of HIV-1. These results indicate that enhanced expression of ARHGDIB renders cells resistant to HIV-1 replication. Consistent with these data, the shRNA-mediated down-regulation of ARHGDIB accelerated the replication of HIV-1 in MT-4 cells (Fig. 1F). These data support the idea that ARHGDIB is a negative regulator of HIV-1 replication.
The RhoGDI family has three members, α, β, and γ, which correspond to ARHGDI A, B, and C, respectively. 11 RhoGDI family members are known to regulate Rho GTPases, including RhoA, Rac1, and Cdc42, although some nonredundant functions of RhoGDIs have been reported. 27 ARHGDIB/RhoGDIβ is primarily expressed in cells of a hematopoietic lineage. Given that Rho GTPases are known to be positive regulators of HIV-1 replication and that ARHGDIB is a negative regulator of Rho GTPases in hematopoietic cells, the function of ARHGDIB was investigated in further detail.
Molecular mediators in the inhibition of HIV-1 replication by ARHGDIB
Under in vitro culture conditions, T cell lines show a constitutively activated cell phenotype, and Rho GTPases, including RhoA, Racl, and Cdc42, have been implicated in T cell activation. 28 –30 These Rho GTPases are expressed in various T cell lines, including MT-4, PM1, and M8166 cells, as confirmed by RT-PCR analysis (data not shown). It seemed likely that augmented ARHGDIB expression affected HIV-1 replication by modulating the activity of these Rho GTPases. Thus, the activation status of RhoA, Rac1, and Cdc42 was determined in relation to the enhanced expression of ARHGDIB.
Rho GTPases are intrinsically inefficient hydrolyzing enzymes that quickly cycle between GTP-bound active and GDP-bound inactive forms. If ARHGDIB attenuates HIV-1 replication by inhibiting Rho GTPase activities, some fraction of Rho GTPases would exist in a GTP-bound active form under steady-state tissue culture conditions, and this population should be decreased in cells ectopically expressing ARHGDIB. Using an active Rho GTPase capture assay, the levels of active Rho GTPases in control and ARHGDIB-expressing cells were measured (Fig. 2A). In this assay, GTP-bound Rho GTPase is captured by glutathione-Sepharose beads conjugated to GST fused to the Rho binding domain of Rhotekin or PAK2. The active forms of both RhoA and Rac1, but very little Cdc42, were detected in PM1 cells under steady-state tissue culture conditions (Fig. 2B–D). Activation levels under the steady-state conditions were estimated by comparing the captured Rho GTPase signals with the maximal activation levels, which were established by converting all Rho GTPases to an active form by GTPγS before the pull-down procedure. The activated fractions of Rac1, RhoA, and Cdc42 were 34.2%, 23.2%, and 0.0%, respectively (Fig. 2B–D). Importantly, ectopic expression of ARHGDIB reduced the activated fractions of RhoA and Rac1 to 4.1% and 3.6%, respectively (Fig. 2B and C). Similar observations were made for MT-4 and SUP-T1 cells (data not shown).
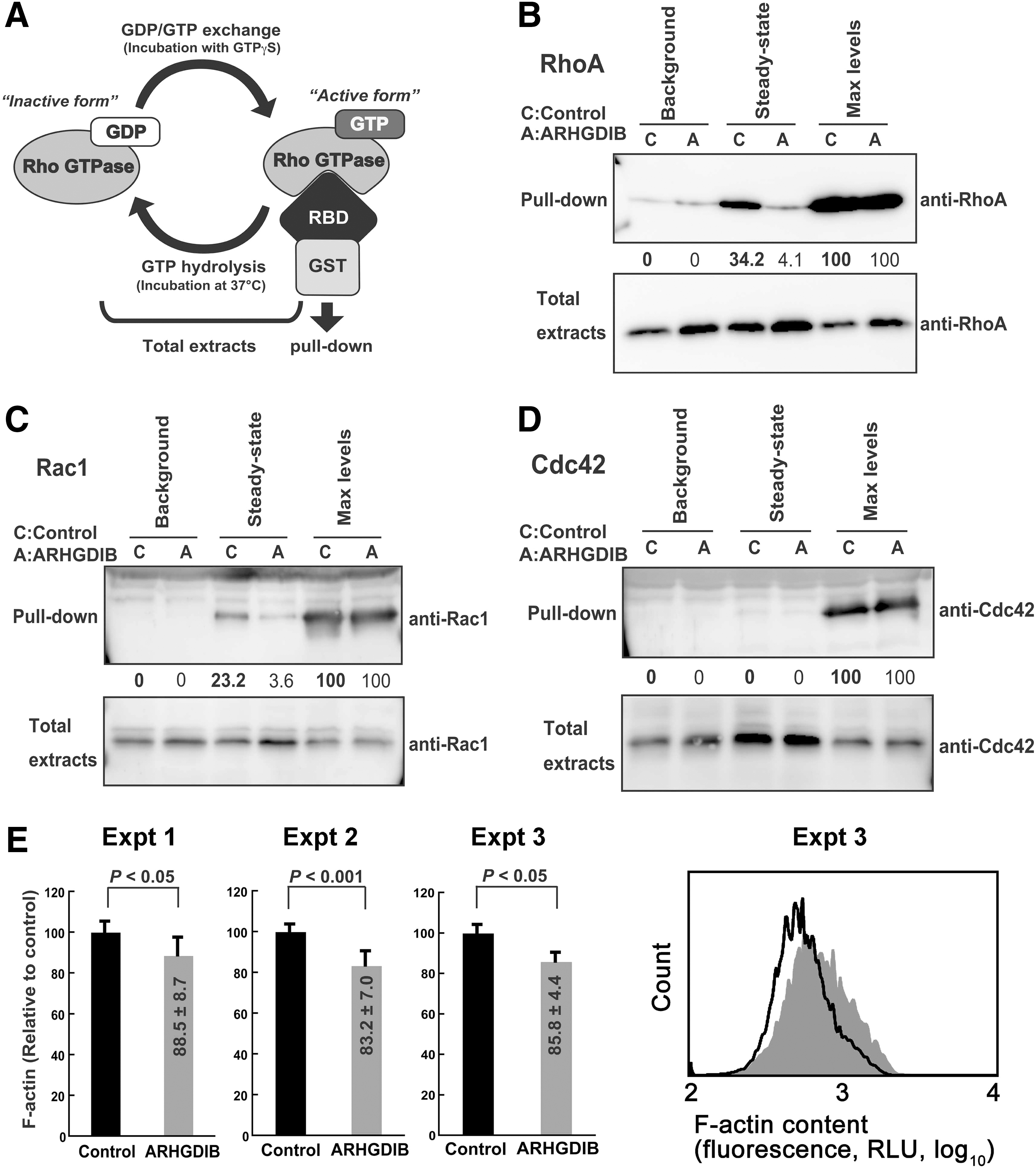
Activation of RhoA and Rac1 under steady-state conditions in PM1 cells.
These observations clearly indicated that ARHGDIB attenuates RhoA and Rac1 activity simultaneously in these T cell lines and suggested that RhoA and Rac1 are the primary targets of ectopically expressed ARHGDIB. Note that RhoA and Rac1 levels were increased by 1.4- and 1.3-fold, respectively, in cells ectopically expressing ARHGDIB, according to the densitometric analysis (Fig. 2B and C). This is due to the ARHGDIB-mediated stabilization of Rho GTPases, as reported previously. 31 This effect is modest in Cdc42 (1.1-fold, Fig. 2D) because the capture of Rho GTPase by ARHGDIB depends on the active GDP/GTP exchange cycle of Rho GTPase under steady-state conditions, which occurs inefficiently in Cdc42.
The common effector function of RhoA and Rac1 is the reorganization of the actin cytoskeleton. 11,12,32,33 If the ectopic expression of ARHGDIB inhibits RhoA and Rac1 effector function, the monomer-polymer actin equilibrium in ARHGDIB-transduced cells should be shifted toward a depolymerized state. 13,14,34 To test this, flow cytometry using fluorescently labeled phalloidin, which binds to F-actin, was used to measure the amount of polymerized actin. Under steady-state conditions, the fluorescence intensity of PM1/ARHGDIB cells was significantly lower than in control cells (Fig. 2E). The reduction levels of F-actin content in PM1/ARHGDIB cells were 14.2±1.6% (N=3, p<0.05 by Student's t-test, two-tailed; Fig. 2E) compared with the control. A similar trend was observed in SUP-T1 cells. These data are in agreement with the results obtained by the active Rho GTPase capture assay and the previous findings that the overexpression of RhoGDI in various cell lines induces the disruption of actin cytoskeleton-dependent processes. 35 –37 These data indicate that the ectopic expression of ARHGDIB in T cells suppresses the activation status of both RhoA and Rac1 under steady-state tissue culture conditions.
Analysis of the viral life cycle in ARHGDIB-expressing T cells
Next, the mechanism by which ARHGDIB blocks HIV-1 replication was investigated. To do this, the viral entry and production phases were examined separately.
To examine the viral entry phase, PM1/ARHGDIB cells were infected with X4-tropic HIV-1NL4-3 Env- or VSV-G-pseudotyped HIV-1 that produces luciferase upon the establishment of infection. In these viruses, luciferase is under the regulation of a long terminal repeat (LTR) or cytomegalovirus (CMV) promoter. When the relative luciferase activity in the control cells was set at 100%, the infection efficiency of HIV-1 Env-pseudotyped HIV-1 expressing luciferase driven by the HIV-1 LTR promoter was 33.0±2.6% (N=3, p<0.001 by Student's t-test, two-tailed; Fig. 3A). Similar results were observed in MT-4 cells (data not shown). In contrast, the infection efficiency of VSV-G-pseudotyped HIV-1 expressing luciferase with either the HIV-1 LTR or CMV promoter was 243.2±31.9% (N=3, p<0.01; Fig. 3A) or 480.3±158.1% (N=3, p<0.05; Fig. 3A), respectively. Similarly, infection with VSV-G-pseudotyped MLV driving the expression of luciferase with the MLV LTR promoter was 10.0±6.4-fold more efficient in PM1/ARHGDIB than in control cells (N=4, p<0.05; Fig. 3A). To examine the effect of ARHGDIB on gene expression, reporter plasmids driving the expression of luciferase with either the CMV or HIV-1 LTR promoter were transfected into PM1 cells. The LTR- and CMV-driven luciferase activities in PM1/ARHGDIB cells were modestly increased compared with the control cells, but not decreased (2.1±0.6-fold, and 2.4±1.6-fold for LTR- and CMV-driven constructs, six and four independent experiments, respectively; Fig. 3B). Taken together, these data suggest that the inhibition of viral entry is specific to HIV-1 Env, and the early phase of the viral life cycle is strongly affected.
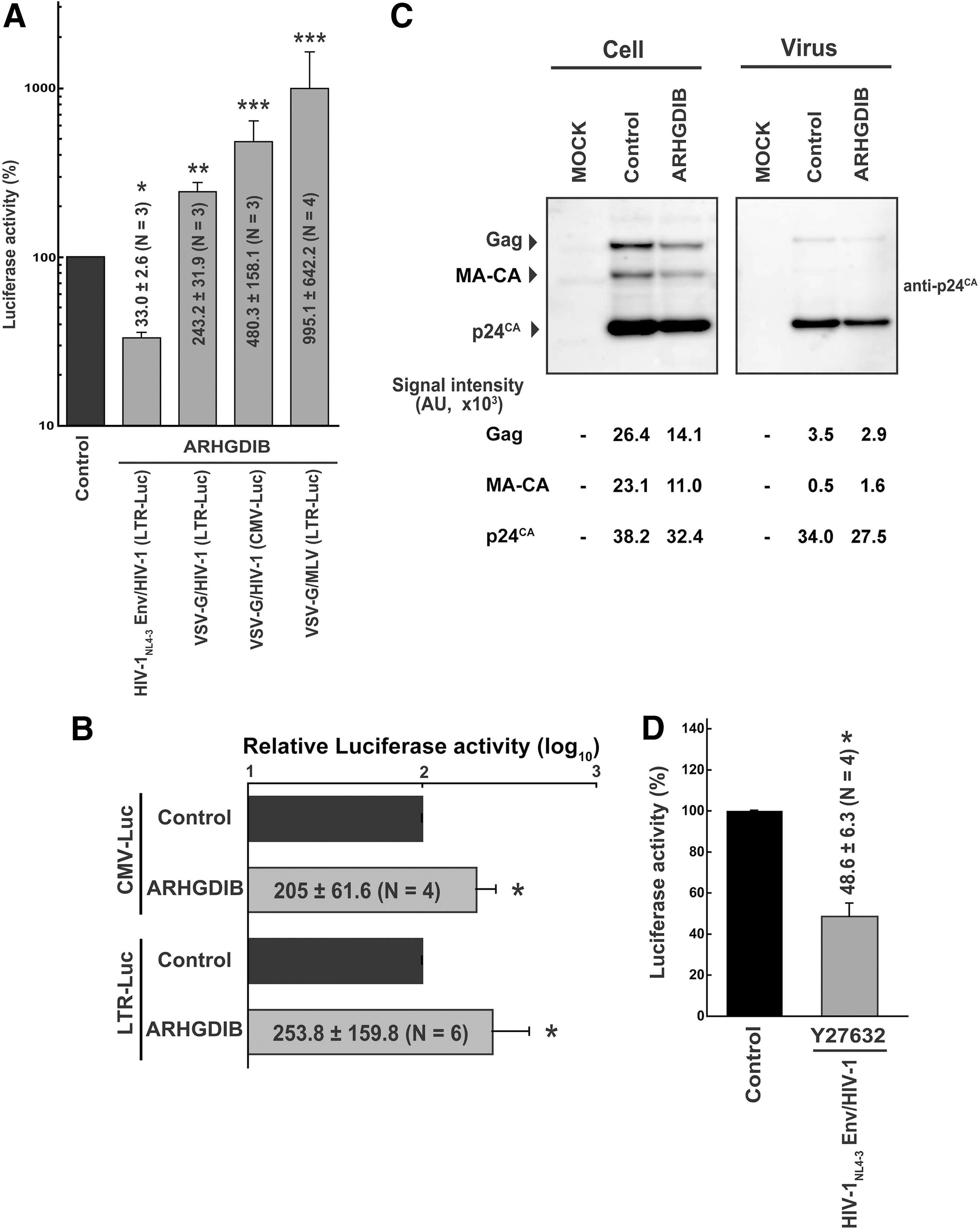
The effect of ARHGDIB expression on the HIV-1 life cycle.
When measured by flow cytometry, PM1/ARHGDIB cells were found to express cell surface CD4 at levels 1.4-fold higher than control cells (average of four independent experiments), while the cell surface levels of CXCR4 were 0.8-fold higher (average of five independent experiments). Considering that ARHGDIB did not affect the levels of cell surface CD4 or CXCR4 in MT-4 cells (Fig. 1B) and M8166 cells (data not shown), and that the reduction in CXCR4 expression in PM1 cells is relatively modest, these data suggest that changes in the expression of viral receptors do not fully account per se for the decreased susceptibility of PM1/ARHGDIB cells to HIV-1NL4-3 Env-pseudotyped HIV-1.
To examine the production phase, proviral DNA was transfected into PM1/ARHGDIB and the control cells, and the viral protein expression in cells and the level of viral production in the culture supernatant were measured. The transfected cells were cultured in the presence of the antiretroviral drug efavirenz, which inhibits the replication cycle of HIV-1, allowing the assessment of viral production in the transfected cells. The levels of viral Gag in PM1/ARHGDIB cells, which are cleaved by protease to yield multiple bands in Western blot analysis with an anti-p24CA antibody, were comparable to the control cells (Fig. 3C). To assess these data quantitatively, densitometric analysis was conducted to quantify the signals representing Pr55Gag, MA-CA, and p24CA. Then, the percentage of p24CA and MA-CA relative to the total signal was calculated; this represented the efficiency of Gag processing. The Gag processing efficiency was 75.5% in PM1/ARHGDIB cells, similar to the control cells (69.9%). In addition, the ratio of the total signal in the viral lysate to that in the cell lysate was calculated. This reflects the efficiency of viral production. The virus/cell intensity ratios were 0.43 and 0.56 for the controls and cells ectopically expressing ARHGDIB, respectively. Similar data were obtained in an independent experiment in PM1 cells. These data suggest that the negative effect of ARHGDIB on the viral production phase was undetectable in T cells.
A single-round HIV-1 infection experiment was performed in the presence of a specific ROCK inhibitor, Y27632, to test whether the effector functions of RhoA are critical in regulating HIV-1 infection. The inhibition of ROCK, a RhoA signal mediator, reduced HIV-1 infection by 48.6±6.3% compared with the control levels (N=4, p<0.05 by Student's t-test, two-tailed; Fig. 3D). These observations suggested that the RhoA signal triggered by HIV-1 Env-receptor interaction is involved in the regulation of HIV-1 infection. This inhibition of ROCK accounted for approximately 77% of the inhibition of HIV-1 infection in PM1 cells ectopically expressing ARHGDIB (33.0% vs. 48.6%; Fig. 3A and D). These data also suggested that Rac1 plays a supplementary role in the restriction of HIV-1 infection. Taken together, this suggests that ARHGDIB limits HIV-1 replication primarily by affecting Env-mediated processes, most likely via receptor clustering and virus-cell membrane fusion.
Discussion
A handful of lymphoid-specific cellular regulators of HIV-1 replication are known, including CD4 and CCR5. However, few hematopoietic lineage-specific inhibitors of HIV-1 replication have been identified. Recently a dendritic- and myeloid-cell-specific restriction factor SAMHD1 has been reported. 38 APOBEC3 family members, which exhibit antiretroviral activity, may be hematopoietic cell specific, although their expression levels in non-T cells have not been directly examined. 39,40 In this study, we showed that modest up-regulation of the hematopoietic cell-specific RhoGDI, ARHGDIB, negatively regulated HIV-1 replication in various T cell lines, while it appeared to have no impact on cell proliferation. ARHGDIB is a constitutively expressed, lymphoid-specific protein. Therefore, ARHGDIB could provide intrinsic immunity against HIV-1 infection. In contrast, APOBEC3 family members are primarily interferon inducible.
RhoGDIs have not been isolated as negative regulators of HIV-1 replication in previous genetic screening studies. This is partly because nonlymphoid cells were used to screen genetic materials and the siRNA/shRNA-based gene knock-down is not able to identify negative regulators of HIV-1 replication at high sensitivity. Moreover, previous screens could not identify all potential factors, especially those present in the hematopoietic cell lineage, since nonlymphoid cells were often used. Our T cell-based screen using replication-competent HIV-1 allowed us to identify ARHGDIB as an HIV-1 inhibitor.
Rho GTPases play multiple roles in cell biology, including actin reorganization, endocytosis, and tubulin regulation, and ARHGDIB is a negative regulator of Rho GTPases. 11 –14,32,41,42 It has been reported previously that inhibition of RhoA affects viral receptor clustering, which lowers the efficiency of virus-cell membrane fusion and thus affects viral replication. 4,43 –45 In contrast, the Rac1-PAK pathway has been shown to support virus-cell membrane fusion. 46,47 Both processes require active actin reorganization. 4,42,44 –47 Interestingly, HIV-1 Env-guided entry is supported by a Filamin A-RhoA-ROCK axis and Arp2/3 complex, both of which are commonly involved in actin cytoskeletal reorganization. 21,44 Our data link the reported functions of RhoA, Rac1, and RhoGDIs with the biological phenotype of ARHGDIB; inhibition of HIV-1 replication in T cells. This HIV-1 inhibitory function of ARHGDIB is exerted via simultaneous functional restriction of two Rho GTPases, RhoA and Rac1. It is likely that receptor clustering and virus-cell membrane fusion are affected in T cells ectopically overexpressing ARHGDIB. As receptor clustering is the initial event of the HIV-1 life cycle, HIV-1 replication is attenuated in such cells regardless of the route of HIV-1 entry. Also, in T cells ectopically overexpressing ARHGDIB, RhoA and Rac1 are substantially inactivated. However, they are not inactivated completely. Thus, these cells are still able to support HIV-1 replication at certain levels, contributing to the delayed HIV-1 replication phenotype in cells ectopically overexpressing ARHGDIB.
Some reports suggest that RhoA inhibits LTR-mediated transcription. 43,48,49 If this is the case, increased ARHGDIB expression should enhance LTR-mediated transcription. In PM1/ARHGDIB cells, HIV-1 LTR-driven luciferase activity was slightly enhanced (Fig. 3B), which is consistent with previously reported findings. On the other hand, HIV-1 replication was repressed in T cells, suggesting that this transcriptional effect on viral replication is modest, if it occurs at all. It is also possible that the enhancement of the luciferase signal may be due to increased endocytic activity in PM1/ARHGDIB cells, since the DEAE-Dextran protocol was employed for transfection, and similar levels of enhancement of CMV-driven transcription were observed in PM1/ARHGDIB cells as well.
Infection with VSV-G-pseudotyped HIV-1 and MLV, which enter cells via endocytosis accompanied by the rearrangement of actin filaments, is augmented in PM1/ARHGDIB cells (Fig. 3A). These findings are consistent with a previous report demonstrating that ARHGDIB stimulates endocytosis in cells with a lymphoid background. 50 The efficiency of VSV-G-pseudotyped MLV infection into PM1/ARHGDIB cells was higher than VSV-G-pseudotyped HIV-1. During the entry phase, HIV-1 utilizes microtubules to traffic toward the nucleus, whereas MLV does not seem to actively do so. 51,52 As ARHGDIB potentially inhibits Rho GTPases, thereby disturbing microtubular organization, it is possible that HIV-1 infection is blocked by ARHGDIB at the microtubule-dependent transport phase in addition to the receptor clustering phase. Alternatively, the relatively high infectious titers produced by the MLV vector may be responsible for the observed phenotype.
Our findings do not negate the possibility that ARHGDIB limits HIV-1 replication by restricting the functions of other effector molecules. For instance, ARHGDIB has also been shown to bind the RhoGEF protein Vav1 and the cytoskeletal protein ezrin, both of which have been implicated in the positive regulation of HIV-1 replication. 53 –57 Thus, these factors may also contribute to the ARHGDIB-mediated inhibition of HIV-1 replication.
The treatment efficacy of HIV-1/AIDS by antiretroviral drugs has greatly improved. However, the adverse effects of antiretroviral drugs lower the quality of life of HIV-1-infected individuals, and the emergence of drug-resistant strains is anticipated for all of the currently available antiretroviral drugs. Insights into HIV-1-host interaction, such as that provided in this study, will aid in the design of novel antiretroviral drugs. Our findings may contribute not only to antiretroviral drug development but also to our understanding of viral pathogenesis. Considering the observation that a modest increase in ARHGDIB expression resulted in the inhibition of HIV-1 replication, it is possible that, at the later phase of HIV-1 infection, CD4-positive cells bearing higher levels of ARHGDIB may be selected. Thus, ARHGDIB levels may be useful as a progression marker of HIV-1 infection. HIV-1 disease progression may be delayed in individuals who have relatively high levels of ARHGDIB in CD4-positive T cells. These possibilities should be examined in future studies.
Footnotes
Acknowledgments
This work was supported in part by the Japan Human Science Foundation, the Japanese Ministry of Health, Labor and Welfare (Research on HIV/AIDS), and the Japanese Ministry of Education, Culture, Sports, Science and Technology (Priority Areas “Matrix of Infection Phenomena” 18073008).
Author Disclosure Statement
No competing financial interests exist.