
Abstract
Mutations associated with the use of protease (PR) and reverse transcriptase (RT) inhibitors have been mostly mapped for HIV-1 subtype B. The prevalence of these mutations in drug-naive HIV-1 subtype B-infected individuals is low but occurs at high frequencies in treated individuals. To determine the prevalence of treatment-associated mutations in non-B viruses, we analyzed a 1613-bp pol region of specimens collected from 57 HIV-1-infected treatment-naive individuals from Cameroon. Of the 57 HIV-1 sequences, 43 belonged to CRF02-AG, two to CRF11-cpx, six to subtype A, one to subtype D, and five were unclassifiable. Of the 57 PR sequences, 100% contained at least one codon change giving substitutions at positions 10, 11, 16, 20, 33, 36, 60, 62, 64, 69, 77, and 89. These substitutions gave the following prevalence pattern, 36I/L (100%, 57/57) >89M/I (98%, 56/57)>69K/R (93%, 53/57)>20I/R (89%, 51/57)>16E (16%, 9/57)>64M (12%, 7/57)>10I (11%, 6/57)>11V (5%, 3/57)=62V (5%, 3/57)=77I (5%, 3/57)>233F/V (4%, 2/57)=60E (4%), which differed significantly from subtype B at positions 20, 36, 69, and 89. All but one (98%) of the 57 RT sequences (438 amino acid residues) carried substitutions located at codons 39A (7%), 43E (7%), 122E (7%), 312Q (2%), 333E (2%), 335C/D (89%), 356K (89%), 358K (14%), 365I (2%), 371V (81%), 376S (11%), or 399D (4%); the frequency of these substitutions ranged from <0.5% to 4% in RT of subtype B. The high prevalence of minor mutations associated with protease inhibitors (PI) and reverse transcriptase inhibitors (RTI) represents natural polymorphisms. HIV-1 PR and RT sequences from antiretroviral (ARV)-naive HIV-infected persons in Cameroon are important for monitoring the development of resistance to PIs and RTIs as such mutations could lead to treatment failures in individuals undergoing ARV therapy.
Introduction
T
Drug resistance threatens the success of ARV treatment programs, especially if unmonitored in areas where there is rapid scale-up of access to ARV drugs. Establishing a baseline database for protease (PR) and reverse transcriptase (RT) sequences from drug-naive HIV-1 nonsubtype B-infected people is important for monitoring and evaluating resistance mutations as was established for subtype B. There is no evidence that natural polymorphisms related with minor resistance mutations or accessory mutations significantly affect drug susceptibility in the absence of major drug resistance mutations, which on the whole are similar in HIV-1 B and non-B subtypes and have not been discovered to be polymorphic. Studies have shown differences in natural polymorphisms associated with PI resistance between PR sequences of HIV-1 subtype B and non-B strains. For example, there are significantly more naturally occurring PI resistance-associated secondary mutations in nonsubtype B than B strains. 6 –8 Furthermore, the patterns of these substitutions have been shown to be different in subtype B strains compared to those in strains of subtypes A, D, C, F, G, and H. 6,7,9 –13 The establishment of a baseline PR and RT sequence database from drug-naive HIV-1 non-B-infected individuals will help inform transmission of resistance viruses and its implication for clinical management. For instance, a high frequency of drug-resistant HIV-1 variants has been reported among recently diagnosed HIV-1 subtype B drug-naive individuals, thus suggesting that alternative drug regimens could be administered. 14 –16
There is disproportionate knowledge of the number of baseline PR and RT sequences from drug-naive HIV-1 subtype B-infected individuals when compared to drug-naive individuals infected with HIV-1 nonsubtype B, a result of a smaller pool of evaluated non-B virus sequences. 4,10,17 –23 Additionally, analysis of drug resistance in RT sequences has in the most part been limited to the first 240 amino acid residues, a region conventionally chosen for drug resistance studies because most NRTI and NNRTI resistance mutations are clustered around the polymerase activity site and its proximal region. 4,17,18,24,25 This precludes previously unreported NRTI treatment-associated substitutions that have been identified at amino acid positions Y318F, G333D/E, N348I, R356K, R358K, and A371V. 26 –30 Additionally, mutations at positions E312Q, G335C/D, N348I, A360I/V, V365I, T369I, A376S, and E399D of the RT connection subdomain together with thymidine analogue mutations are associated, in subtype B, with enhanced clinical resistance to zidovudine. 29,31,32 Thus, additional knowledge gained by analyzing larger RT sequences would help our understanding of drug resistance-associated mutations for non-B virus strains.
In the present study, we describe the genetic diversity in PR and RT (spanning the first 438 amino acid residues of RT) sequences in drug-naive individuals infected with HIV-1 non-B subtypes, and analyze for the presence of polymorphisms that, for subtype B, are mutations associated with drug resistance.
Materials and Methods
Specimens
Between 2002 and 2003, blood was collected from 57 HIV-1-positive individuals from Cameroon. These included 21 HIV-positive blood donors (anonymously unlinked) from urban cities of Yaoundé and Douala, and from 36 individuals living in rural areas: Banga (n=23), Banso (n=3), Mbingo (n=5), Mamfe (n=3), and Nguti (n=2). To our knowledge, none of these persons had received any ARV treatment. Antibodies to HIV were detected by ELISA using the HIV-1/HIV-2 EIA kits (Murex or Genetic Systems Corporation, Redmond, WA) and confirmed by Western blot (Immunetics Qualicode HIV-1/2, Boston, MA). The specimens were collected under the HIV genetic variant protocol #1367 approved by the CDC Institutional Review Board and the Ethical Committee of the Ministry of Health of Cameroon.
DNA extraction, amplification, sequencing, and genetic analyses
Proviral DNA was extracted directly from peripheral blood mononuclear cells (PBMCs) with the Generation Capture Columns (Generation Inc., Minneapolis, MN) and used for nested polymerase chain reaction (PCR) amplification of an approximately 1.613-kb pol fragment comprising the entire PR of 299 bp and the first 1.314 kb pairs of RT with the following primers: outside DP10-forward (5′-CAACTCCCTCTCAGAAGCAGGAGCCG-3′) and RT11-reverse (5′- CTAATGCATACTGTGAGTCTGTTACTA-3′) and nested DP16a-forward (5′- CCCTCAAATCACTCTTTGGCA-3′) and RT17-reverse (5′- CTGCCCCATCTACATAGAAAGTTTC-3′). The amplification reaction was performed with Platinum Taq DNA Polymerase High Fidelity PCR system (Life technology, Bethesda, MD) according to the manufacturer's instructions. Briefly, the PCR conditions for the first and the nested rounds were the same and included a denaturation step of 2 min at 94°C, followed by 35 cycles of 30 s at 94°C, 30 s at 55°C, and 60 s at 72°C with a final extension of 3 min at 72°C. Amplified PCR products were purified using the PCR purification kit (Qiagen, Avenue Stanford, Valencia, CA) and directly sequenced with automated DNA sequencer ABI model 377 (Applied Biosystems, Foster City, CA) using nested PCR primers along with others if needed [DP11-reverse: 5′-CCATTCCTGGCTTTAATTTTACTGCTA-3′; primer E-reverse: 5′-CCATCCCTGTGGAAGCACATTG-3′; and primer D-reverse: 5′-TCTGTATGTCATTGACAGTCCAGC-3′ (1); AY1680-forward: 5′-ATGACAAAAATCTTAGAGCCCTTTAGA-3′; and RTF3-forward: 5′-GAAGCAGAATTAGAATTGGCAGAGAA-3′]. The derived nucleotide sequences were aligned using the CLUSTAL W multiple sequence alignment program with the reference strains of groups M, N, and O obtained from the HIV-1 Los Alamos database (
Amino acid analysis
The aligned DNA sequences were translated to amino acids (aa) using the Genetic Data Environment (GDE) package.
34
The first seven aa of the PR gene that are part of the 5′ PCR primer DP16a were excluded from analysis. Non-B subtype amino acid sequences were aligned and compared to the consensus B sequence. Information on the major, minor, and connection subdomain mutations associated with PI and RT resistance was obtained from the literature (
Polymorphism definition
Polymorphisms of ART-associated mutations were defined as substitutions that occurred in more than 1% of sequences from untreated persons. Non-B-specific polymorphisms (subtype-signature substitutions) was characterized as substitutions that were significantly more prevalent (>60%) in non-B subtype than B viruses from drug-naive persons. 17,18 The p value for the resultant comparison was calculated using Fisher's exact test.
Results
Phylogenetic subtypes and genetic diversity of DNA sequences
Phylogenetic analysis of 1613-bp pol fragments comprising the PR and first 1314 bp of the RT gene classified unambiguously 52 of the 57 sequences into distinct HIV-1 group M subtypes or CRFs (Fig. 1): 43 clustered with CRF02-AG sequences, six belonged to subtype A, two were CRF11-cpx, and one was subtype D. The remaining five unclassifiable sequences belonged to HIV-1 group M but did not significantly cluster (bootstrap value >70%) with any known subtypes or CRFs in this region. Subtype distribution patterns were similar between the 36 HIV-1 strains from the rural areas and the 21 HIV-1 strains collected from blood donors in cities. For example, CRF02-AG strains were predominant in both rural and blood donor specimens, 88% and 62%, respectively. Although more CRF02 sequences were from rural areas (29/36) when compared to urban regions (13/21), these differences were not statistically significant (p=0.22, Fisher's exact test).
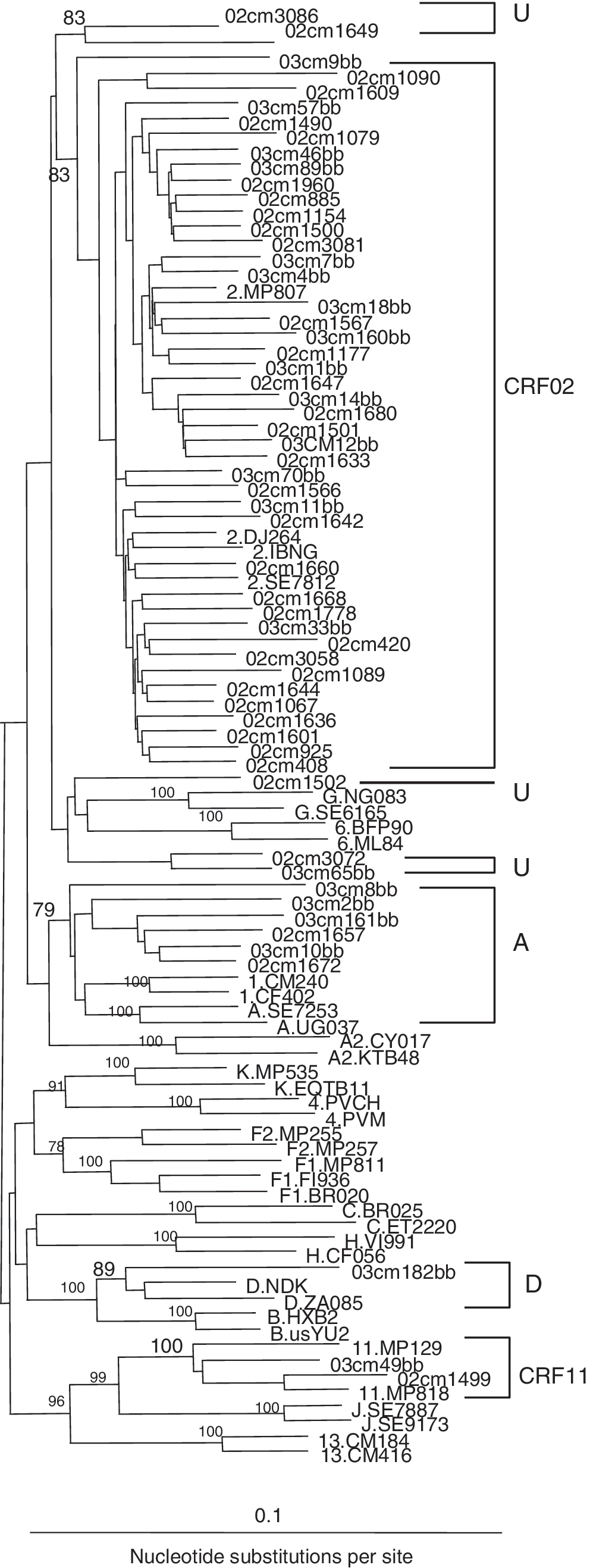
Phylogenetic classification of 57 Cameroonian HIV-1 pol sequences (1613 bp each) comprising the protease (PR) and the first 1314 bp reverse transcriptase (RT) gene regions; they are preceded by the prefix ’02 and 03 cm’ indicating the year (2002 and 2003, respectively) and the country (Cameroon) of specimen collection. The 21 sequences ending with “bb” were from urban blood bank donors; the other 36 sequences were from the rural areas. HIV-1 references for subtypes A–D, F–H, J, and K of group M are preceded by capital letters; CRFs (CRF01, CRF02, CRF04; CRF06, CRF11, and CRF13) are preceded by numbers (1, 2, 3, 4, 6, 11, and 13, respectively). Clustering of Cameroonian sequences with subtype/CRF is delineated; U indicates unclassifiable sequences. The numbers at the nodes correspond to the bootstrap values; only values >70% are shown. The scale bar indicates an evolutionary distance of 0.1; vertical distances are for clarity only.
Sliding-window bootscan analysis of five unclassifiable PR-RT sequences showed a similar recombinant pattern in two sequences consisting of subtype G in PR and subsubtype F2 and subtype A in the RT (data not shown). Furthermore, phylogenetic analysis confirmed these findings and also revealed that subtypes PR-G and RT-A originated from CRF02-AG as they significantly clustered with CRF02-AG reference strains when analyzed together (data not shown). The other three unclassifiable strains represented different recombinants consisting of PR subtype G (CRF02-AG like) in combination with different fragments of subtypes A, G, K, F, or unclassifiable in the RT sequence (data not shown).
PI treatment-associated mutations
To examine the frequency of minor non-B subtype PI-resistance-associated substitutions, 21 aa positions were analyzed within the PR region (L10I/F/V/C, V11I, G16E, K20R/M/I/T/V, L24I, L33I/F/V, F34Q, M36I/L/V, K43T, F53L/Y, D60E, I62V, L63P, I64L/M/V, H69K/R, A71V/I/T/L, G73C/S/T/A, V77I, I85V, L89M/V/I, I93L/M), which are associated in vivo with HIV-1 exposure to tipranavir, saquinavir, ritonavir, indinavir, nelfinavir, fosamprenavir, atazanavir, darunavir, or lopinavir in subtype B viruses. 36 Of the 57 Cameroonian sequences, 100% (57/57) contained at least one substitution at codon 10, 11, 16, 20, 33, 36, 60, 62, 64, 69, 77, and 89 (Fig. 2). These substitutions gave the following prevalence pattern: 36I/L (100%, 57/57)>89M/I (98%, 56/57)>69K/R (93%, 53/57)>20I/R (89%, 51/57)>16E (16%, 9/57)>64M (12%, 7/57)>10I (11%, 6/57)>11V (5%, 3/57)=62V (5%, 3/57)=77I (5%, 3/57)>233F/V (4%, 2/57)=60E (4%).
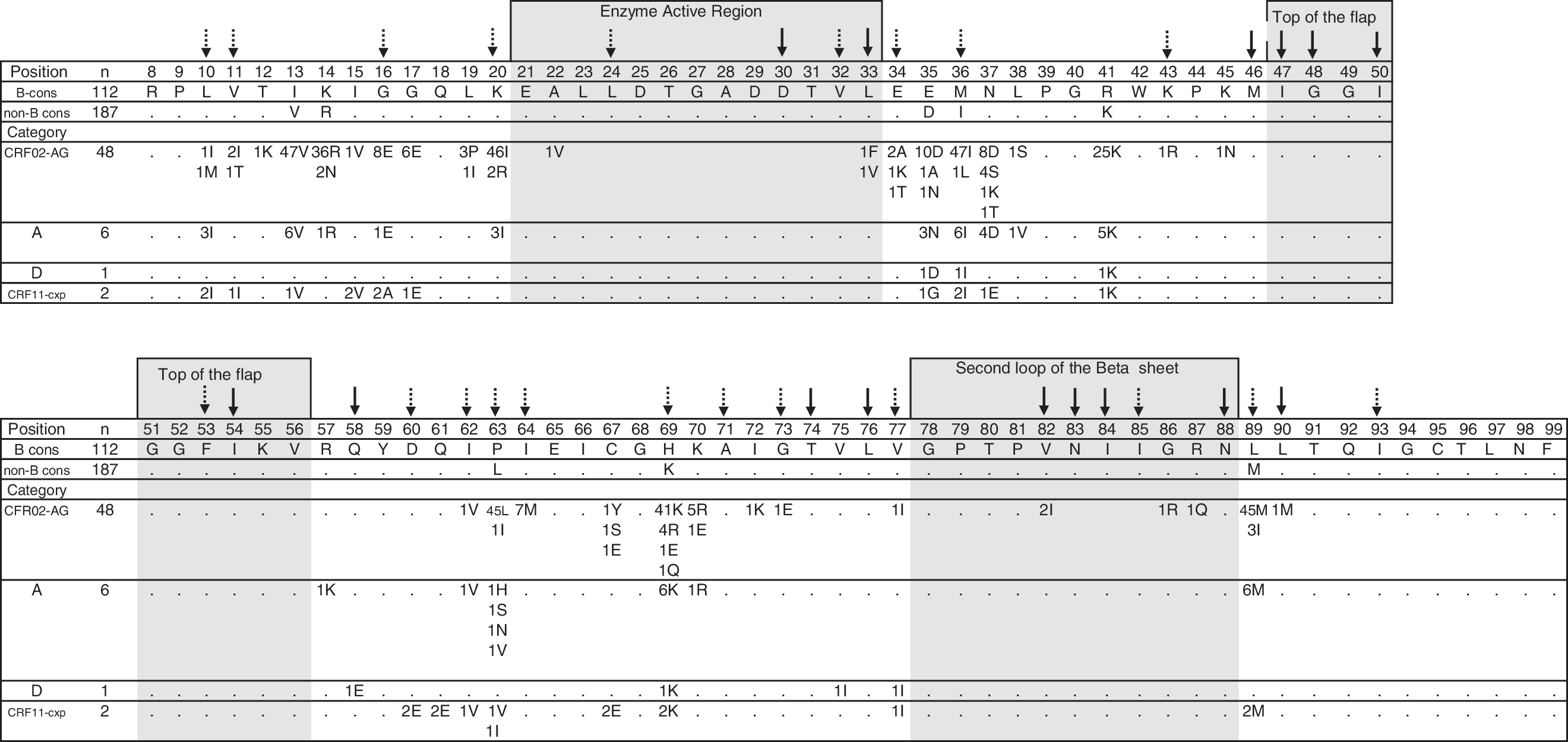
Amino acid polymorphisms of HIV-1 PR sequences isolated from PI-naive Cameroonian individuals infected with HIV-1 CRF02-AG, subtypes A and D, and CRF11-cpx. The number of strains that possessed a particular substitution is shown below each amino acid position. When the number does not total the number of sequences analyzed, the difference reflects an amino acid of subtype B consensus sequence obtained from the Los Alamos database (
When the frequency of PI-associated substitutions in Cameroonian non-B PR sequences was compared with those reported for subtype B PI-naive strains,
37
the prevalence was significantly higher (p<0.01) at positions 36I/L, 89M/I, 69K/R, and 20I/R among non-B (100%, 100%, 93%, and 89%, respectively) than in B viruses
Our data revealed one major mutation, 90M, in one individual infected with CRF02-AG when 15 major mutation positions were analyzed (D30N, V32I, M46I/L, I47V, G48V, I50L/V, I54M/L, Q58E, T74P, L76V, V82A/F/T/S, N83D, I84V, N88S, and L90M). By contrast, minor resistance-associated substitutions (L10I, V11I, G16E, K20I/R, L33F, M36I, D60E, I62V, I64M, H69K/R, V77I, and L89M/I) were found in all Cameroonian PR sequences regardless of subtype or CRF (Fig. 2). Of these, all strains harbored dual (68%, 39/57) or triple (22%, 13/57) PI-associated mutations, while only one (2%) strain carried a single PI minor mutation (Fig. 2). Also, our data indicate that whereas the majority of Cameroonian PR CRF02-AG sequences carried either a combination of 20I/36I (96%, 46/48) alone or with 69K or 89M mutations, the sequences of the subtypes A and D and CRF11-cpx harbored mainly 10I/36I alone or with 69K or 89MI substitutions, which is similar to previous findings thus suggesting that the PI mutational patterns of PR CRF02-AG sequences may differ from patterns of other nonsubtype B viruses. 7
RTI treatment-associated substitutions
We analyzed the RT sequences for the presence of 20 previously unreported drug treatment-associated substitutions (T39A, K43E/Q/N, K122E, H208Y, D218E, H221Y, L228H/R, E312Q, Y318F, G333D/E, G335C/D, N348I, R356K, R358K, A360I/V, V365I, T369I, A371V, A376S, and E399D). 5,29 –32 Our analyses revealed these substitutions to be present at 12 positions within Cameroonian sequences: 39, 43, 122, 312, 333, 335, 356, 358, 365, 371, 376, and 399 (Fig. 3). Their frequencies were <14% for T39A (7%, 4/57), K43E/Q/N (7%, 4/57), K122E (7% 4/57), E312Q (2%, 1/57), G333E (2%, 1/57), R358K (14%, 8/57), V365I (2%, 1/57), A376S (11%, 6/57) and E399D (4%, 2/57), whereas they were >80% for G335C/D (89%, 51/57), R356K (89%, 51/57), and A371V (81%, 46/57). Of these, more than 95% (55/57) of strains carried at least two substitutions. The dual pattern 356K/371V was the most common (75%, 43/57), followed by other combinations (15%, 9/57).
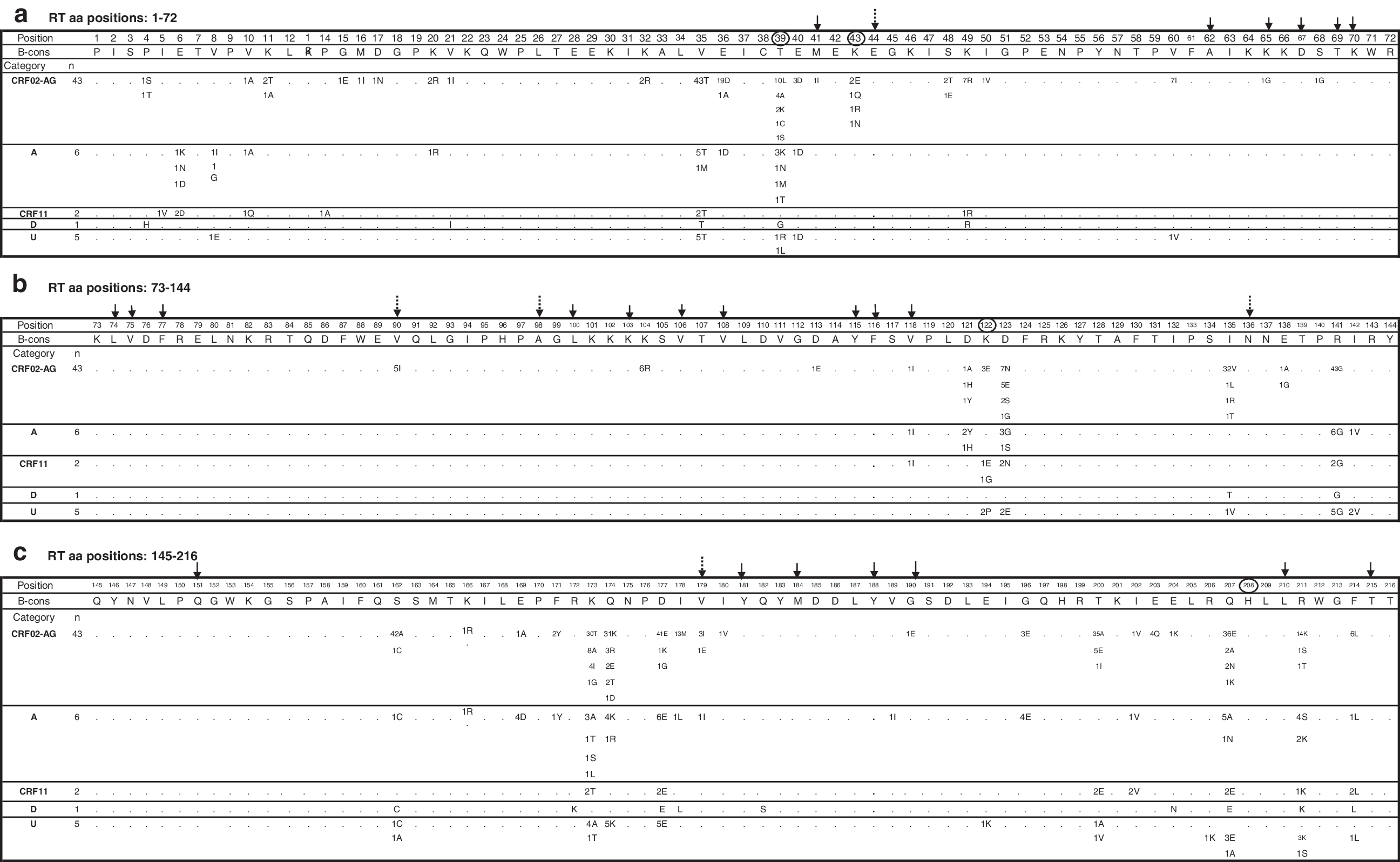
Amino acid polymorphisms of HIV-1 RT sequences isolated from RTI-naive Cameroonian individuals infected with HIV-1 CRF02-AG, subtypes A, D, J (CRF11-cpx), and unclassifiable strains. The number of strains that possessed a particular substitution is shown below each amino acid position. When the number does not total the number of sequences analyzed, the difference reflects an amino acid of subtype B consensus sequence obtained from the Los Alamos database (

To examine the frequency of known minor and major RTI resistance-associated substitutions, 33 amino acid sites were analyzed including 18 NRTI-associated positions (M41L, E44D, A62V, K65R, D67N, T69D, K70R, L74V, V75I, F77L, Y115F, F116Y, V118I, Q151M, M184V, L210W, T215Y/F, and K219Q/E) and 15 NNRTI-associated sites (V90I, A98G, L100I, K101P/E/H, K103N, V106M/A/I, V108I, E138A/G/K, Y181C/I/V, Y188L/C/H, G190S/A/E, P225H, M230L, P236L, and K238T). This analysis showed that 12.3% (7/57) of HIV-1 Cameroonian strains carried single mutations that for subtype B are associated with RTI resistance (Fig. 3). Briefly, three strains (CRF02, CRF11, and subtype A) harbored substitution V118I that if combined with E44D is associated with NRTI exposure and might confer low level resistance to zidovudine or lamivudine, one subtype D strain had mutation G333E that may facilitate AZT resistance in the presence of the M184V substitution, and one CRF02 strain carried a rare G190E substitution that could contribute to resistance to efavirenz, nevirapine, and delaviridine.
Discussion
Our study evaluated treatment resistance-associated mutations in PR and an expanded spectrum of RT sequences (including the connection subdomain of RT) of drug-naive HIV-1 non-B-infected individuals. We show first that the frequencies of these polymorphisms in the PR regions of non-B HIV were similar to B in all but four sites, positions 20, 36, 69, and 89, where the frequency was higher in non-B viruses than in B viruses. Second, for the RT region, the frequency of polymorphisms was generally higher at three sites (335C/D, 356K, and 371V) in the connection subdomain (amino acids 316–437) for non-B viruses compared to B viruses. Third, the prevalence of major mutations associated with resistance to PI, NRTI, and NNRTI in nonsubtype B was very low (only one, L90M in PR and G190E in RT, respectively).
Although HIV-1 subtypes other than B are responsible for most infections worldwide, drug resistance sequence data are limited for individuals infected with non-B viruses, and there is evidence to suggest differences in antiretroviral drug resistance to different classes of drugs in non-B isolates. For example, there is a predominance of L90M rather than D30N among persons infected with non-B viruses exposed to nelfinavir. 5 Furthermore, isoleucine instead of valine in position 82 (a site of major resistance to PIs) is significantly more prevalent in some non-B viruses and may decrease susceptibility to PIs. 38 Also, drug-naive individuals infected with non-B viruses were shown to have a greater number of tipranavir-associated mutations than B viruses. 39 While polymorphisms occur in both the PR and RT region in drug-naive individuals, some of the amino acid substitutions occurred at higher rates in non-B virus at positions associated with drug resistance in subtype B. 5
The analyses of the PR and a larger RT (438 amino acid residues) region enabled us to analyze these mutation sites (characterized for subtype B), in particular, mutations at the connection subdomain RT region that would otherwise have been undocumented by just looking at the traditional RT region comprising 240 amino acid residues. Our study revealed a strikingly higher prevalence (>80%) at three of the 20 sites characterized for subtype B. These sites, 335C/D, 356K, and 371V, were located in the connection subdomain and may represent non-B subtype-specific substitutions. Minor mutations in the connection subdomain such as E312Q, G335C/D, N348I, A360I/V, V365I, A371V, and A376S in the presence of TAMs have been shown to increase resistance to zidovudine from 11-fold to as much as 536-fold over wild-type RT.
31,32
Also mutations (N348I, T369I, E399D) at the connection subdomain of RT have been associated with resistance to zidovudine and NNRTIs.
29,31
The substitutions in unclassifiable sequences revealed a similar distribution as in subtype-assigned Cameroonian strains. This suggests that recombination processes that occurred within the RT region may not affect natural polymorphisms of ARV-associated mutations. In contrast to Cameroonian findings, the frequency of these substitutions in drug-naive RT from subtype B viruses was low at all positions analyzed and ranged from <0.5% to 4% (
Overall, we identified a high frequency of PR polymorphisms related to mutations associated with drug treatment resistance in non-B strains of Cameroonian origin, which showed different mutational patterns between non-B and B viruses. The impact of these mutations in treated patients infected with non-B viruses is not known. Similar to the RT region, these mutations in the PR region occur alongside known major mutations. For instance, an increased frequency of mutations at positions K55R, I85V, C95F (indinavir treated), K45R, T74S (neflinavir treated), E34Q, K53T (lopinavir/ritonavir treated), and K55R (lopinavir/ritonavir and nelfinavir) was seen in subtype B-treated patients and all were involved with one or multi-PI resistance-associated mutations at positions 82, 84, and 90. 41
Only one major PI resistance-associated mutation (90M) and one major RTI resistance-associated mutations (G190E) were observed, and consistent with previous studies indicating major PI or RTI resistance-associated mutations in drug-naive individuals are of low prevalence. 4,7,8,10,16,42,43 Our data showed a high prevalence of multiple secondary PI-associated mutations (68% dual mutations) and only 2% for single PI minor mutations. These data seem to contrast with our previous findings showing that 38% of Cameroonian strains harbored multiple PI substitutions, whereas 62% carried single mutations although the subtype distribution was similar in both cases. 10 The difference could partly be accounted for by the fact that in our present study, the 20I mutation occurred in 86% (49/57) of Cameroonian sequences (mainly in CRF02-AG) (Fig. 2). The 20I mutation has previously been reported in PR of CRF02-AG strains in West Africa including Cameroon, 8,10,43 but it was only recently classified as the secondary mutation that confers resistance to lopinavir, thus contributing to the discrepancy observed. 44,45
Universal access to ARV treatment is being scaled up in developing countries. In Cameroon, this goal took a significant step forward in 2007 when the ministry of health declared as part of its national program that all antiretrovirals would be made available to all eligible individuals. This effort will inevitably lead to increased access and use of ARVs; thus, monitoring for the emergence of resistance into the population is important and a proper understanding of these mutations and their patterns can be gained only against a well-established baseline database from infected drug-naive individuals.
In summary, our study provides valuable baseline database for non-B subtype viruses from drug-naive individuals including analyses of previously unreported treatment-associated mutations identified for subtype B viruses. The identification of a higher prevalence of a few PI- and RTI treatment-associated substitutions in non-B relative to subtype B viruses further documents genetic differences between these two categories of strains.
Sequences
The GenBank accession numbers for CRF02-AG sequences are DQ166383–DQ166426; for CRF11-cpx DQ166427–DQ166428; for subtype A DG166429–DQ166434; for subtype D DQ166435; and for unclassifiable DQ166436–DQ166440.
Footnotes
Acknowledgments
The authors would like to acknowledge Gerardo Garcia-Lerma for scientific advice and critical review of the manuscript.
Author Disclosure Statement
No competing financial interests exist. The findings and conclusions in this report are those of the authors and do not necessarily represent the views of the Centers for Disease Control and Prevention. The use of trade names is for identification only and does not constitute endorsement by the United States Department of Health and Human Services, the Public Health Service, or the Centers for Disease Control and Prevention.