
Abstract
Recombination contributes substantially to the genetic diversity of HIV-1, and is likely to occur in populations in which multiple subtypes circulate. Molecular epidemiological studies showed that subtype B, CRF01_AE, and CRF07_BC are currently circulating in parallel among men who have sex with men (MSM) in China, suggesting the possible emergence of new recombinants. In the present study, we identified two new HIV Type 1 unique (CRF01_AE /B) recombinant forms in this population by near full-length genomic analysis. Our data provided the first description of the near full-length genomes of these new CRF01_AE/B recombinants as well as important insights into the complexity of HIV-1 recombinant strains currently in circulation among MSM in China. These data highlight the importance of continuous surveillance of the dynamic change of HIV-1 subtypes and new recombinants among the MSM population.
HIV-1
Recombination substantially contributes to the overall genetic diversity of HIV-1, and mosaic strains usually arise in populations in which multiple subtypes are circulating. It is observed that multiple HIV-1 genotypes, including subtype B, CRF01_AE, and CRF07_BC, are currently circulating in the population of men who have sex with men (MSM) in China, 2 and the prevalence of HIV-1 among MSM has been precipitously increasing from 12.2% in year 2007 to 32.5% in year 2009 in newly identified HIV-1-infected cases in China. 3 The cocirculation of multiple HIV-1 genotypes and rapid spreading could facilitate the generation of new recombination forms. Indeed, we identified and characterized two novel recombinants derived from subtype B and CRF01_AE, which were highly different from the previously reported CRF01_AE/B recombinant. 4 –7
The study was approved by Institutional Ethic Committee and written informed consents were obtained from study subjects. Plasma samples were collected from two HIV-1-infected MSM (HIV+MSM) who were identified as complex recombinant HIV-1 infection in our routine molecular epidemiological screening. Detailed information concerning the two patients is listed in Table 1. Both of the patients were infected through homosexual contact and confirmed as HIV-1 seropositive in December 2006 for 08CYM015 and in April 2007 for 08CYM047, respectively. Subject 08CYM015 claimed to be exclusively homosexual, had more than 100 male sexual partners in his life including several German partners, and engaged in group sex four times. In the past 3 months, he reported having anal sex with 10 men and preferred receptive anal intercourse. He was gonorrhea positive. Subject 08CYM047 also claimed to be exclusively homosexual and had 10 male sexual partners in his lifetime. In the past 3 months, he reported having anal sex with one man and preferred insertive anal intercourse. He was condyloma accuminata positive. CD4+ T cell counts were 532 cells/μl and 402 cells/μl and viral loads were 7700 copies/ml and 780,700 copies/ml for 08CYM015 and 08CYM047, respectively.
For full-length genome sequencing, RNA was extracted from a 1.0 ml plasma specimen as instructed by the column purification method (QIAamp Viral Mini Kit, Qiagen, Hilden, Germany). Virions were initially concentrated from the plasma by centrifugation at 21,000×g for 120 min at 10°C. All but 140 μl of the supernatant was removed before lysis buffer was added. Extracted RNA (50 μl) was reversely transcribed into cDNA using Superscript III (Invitrogen, USA) with primer UNINEF7 or VIF-VPUoutR1, and three regions of the viral genome, corresponding to nucleotides 623–3338 (5'-end 2.7 kb), 2483–6231 (the middle 3.8 kb), and 5861–9181 (3'-end 3.3 kb) in HXB2, were independently amplified from cDNA by using nested polymerase chain reaction (PCR) to contain a nearly full-length genome of HIV-1(amplification primers listed in Table 2). 8 All PCRs were performed with Takara ExTaq Hot Start Version (TaKaRa Biotechnology Co., Ltd., Dalian, China).
(+) Sense primer; (−) antisense primer.
Positions according to the HXB2 numbering system (GenBank accession number K03455).
Sequence: R (purine: A and G) or Y (pyrimidine: T and C).
The same conditions of amplification were used in two rounds for three amplicons: 94°C for 2 min; 10 cycles of 94°C for 10 s, 60°C for 30 s, 68°C for 4 min; 20 cycles of 94°C for 10 s, 55°C for 30 s, 68°C for 4 min; with a final extension at 68°C for 10 min. The positive PCR products were separated by agarose gel electrophoresis and purified by using a Qiagen Gel Extraction Kit (QIAGEN Inc., Germany). Purified PCR products were directly sequenced with an ABI Prism Big Dye Terminator Cycle Sequencing Reaction Kit (Applied Biosystems) and the ABI model 377 automated sequencer. Sequencing primers were designed either by gene walking or against conserved regions of full-length reference genomes and are available on request. The nearly full-length genome (NFLG) sequence (8.5 kb: from nucleotide 649 to nucleotide 9181 in HXB2) was assembled by overlapping the sequences of three amplicons and merging them into one sequence as the overlapped sequences from two fragments contain greater than 98% homology. Consequently, all gene sequences except nef were successfully obtained.
To locate the open reading frames (ORFs) for each of the relevant genes, manual adjustment was carried out on the alignment using BioEdit (
In the phylogenetic trees constructed with full genome reference sequences (Fig. 1), the 08CYM015 sequence was clustered with subtype B reference sequences but supported by a weak bootstrap value (67%). In contrast, the 08CYM047 sequence was clustered with CRF01_AE, CRF33_01B, and CRF34_01B reference sequences with a high bootstrap value (94%). Furthermore, similarity plot analysis was performed using reference strains of subtype A1, B, C, H, and CRF01_AE to prove the recombinant structure after removing all gaps in the alignment using the online Gapstreeze software (
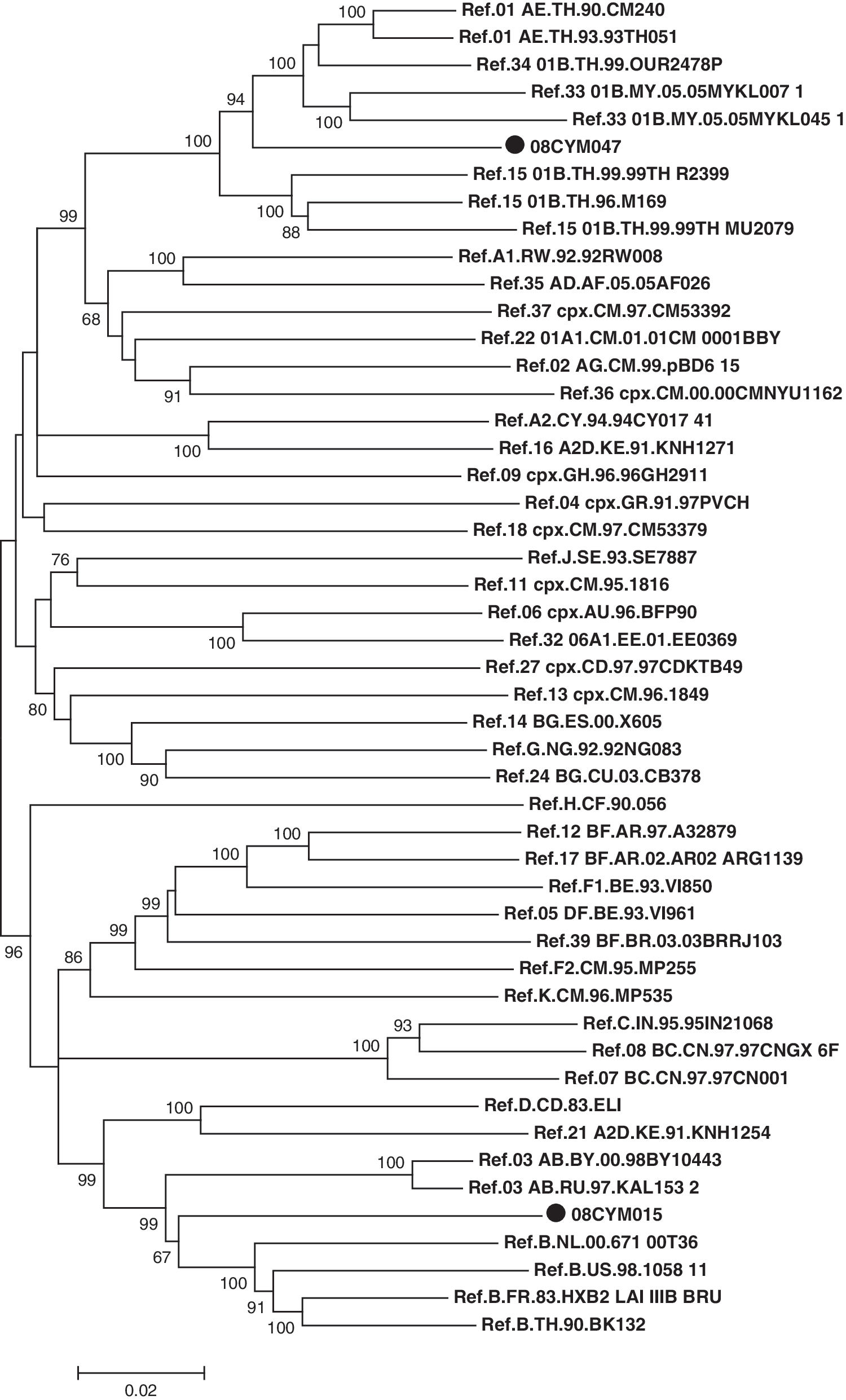
Neighbor-joining phylogenetic analysis of near full-length sequences of 08CYM015 and 08CYM047. The indicated sequences (marked with a black circle) and HIV-1 subtype reference sequences available in the Los Alamos database were collected to construct a phylogenetic tree by using CLUSTAL W with minor manual adjustments. The statistical robustness of the neighbor-joining tree and reliability of the branching patterns were confirmed by bootstrapping (1000 replicates). Bootstrap values >65% were shown at the nodes of the tree. Horizontal branch lengths were drawn to scale.
In contrast, the backbone of the 08CYM047 genome was CRF01_AE with three subtype B fragments inserted in the pol and nef region (Fig. 2). To identify putative recombination breakpoints, both the near full-length genome sequences were analyzed by bootscanning using SimPlot software. They were aligned with HIV-1 subtype A1, B, C, H, and CRF01_AE reference sequences and used for bootscanning after the gap was stripped by using a window size of 300 nucleotides, a step size of 20, the Kimura (two-parameter) distance model, and the neighbor-joining method; the transition/transversion ratio was set as 2.0. Although the backbone of the 08CYM015 genome was the B subtype, two CRF01_AE fragments were identified to insert into the subtype B backbone at the location of nucleotide position 6738–6991 and 7355–9031, respectively, as referred to the HV-1 prototype HXB2 genome, which was further proved by FindSites analysis (Fig. 3). Interestingly, 08CYM047 employed CRF01_AE as its backbone and three subtype B fragments were inserted at the location of nucleotide positions 2752–3404, 3815–4618, and 8939–8995, respectively, as referred to the HXB2 complete genome (Fig. 3).
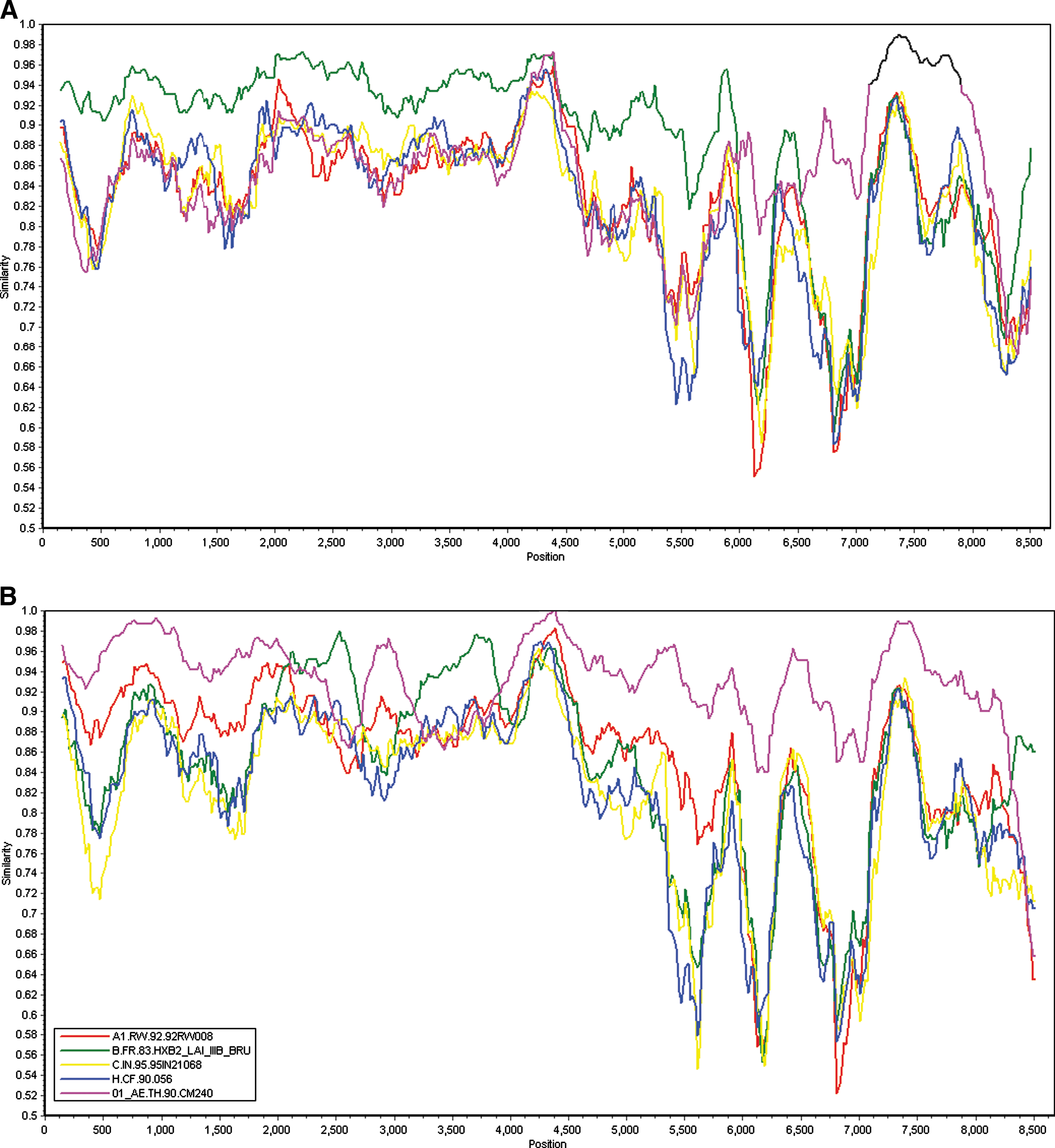
Similarity plots of 08CYM015 and 08CYM047 against reference strains. Similarity plots of 08CYM015

Bootscan analysis of the near full-length nucleotide sequences of 08CYM015
To demonstrate the genomic graphic structure, the Map-Draw Tool available at the Los Alamos HIV sequence database was used to generate the genomic map of 08CYM015 and 08CYM047 (Fig. 4). It is shown again that two CRF01_AE segments were inserted into the backbone of the subtype B strain at the env, tat2, rev2, and nef regions in the 08CYM015 genome and three subtype B segments were inserted into the backbone of the CRF01_AE strain at the pol and nef regions in the 08CYM047 genome. Although CRF01_AE/B viruses have been described in the literature, 4 –7 the mosaic composition of these isolates differs from those of strains 08CYM015 and 08CYM047, hence they were classified as URF_B 01 and URF_01B, respectively.
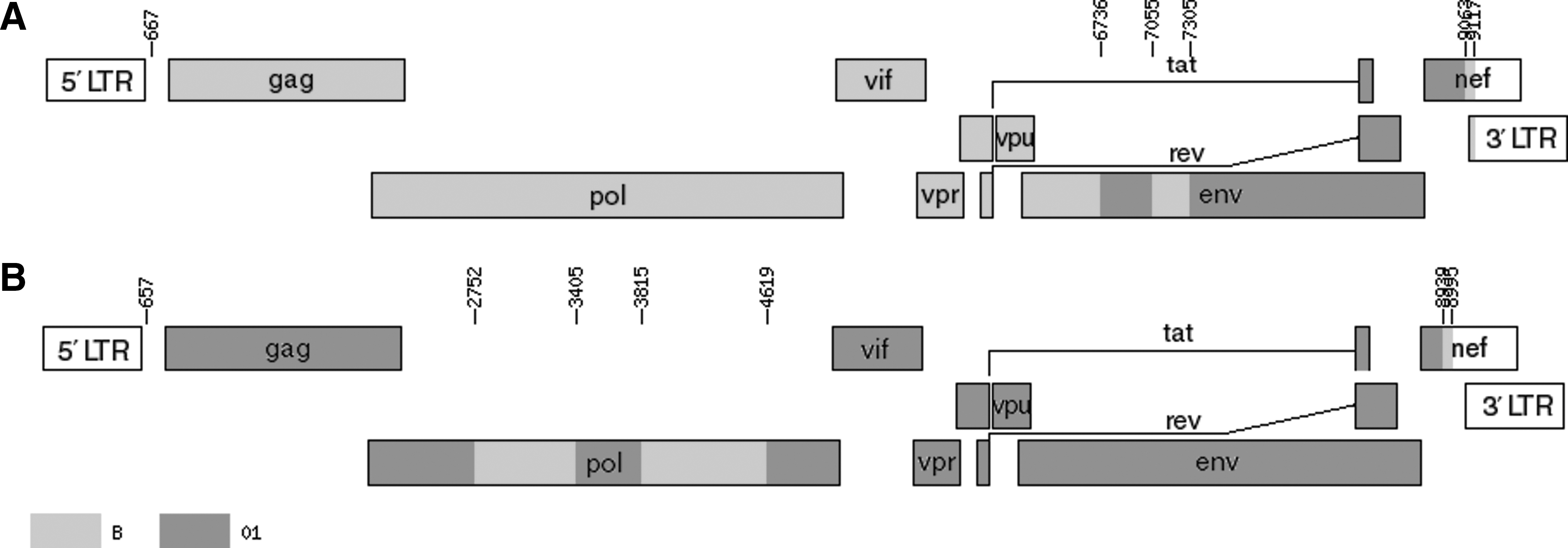
Schematic illustration of subtype structures of 08CYM015 and 08CYM047. Map-Draw Tool available at the Los Alamos HIV sequence database was employed to generate the mosaic structure of 08CYM015 and 08CYM047.
Subtype B and CRF01_AE have been circulating in China since the 1990s, predominantly among MSM for subtype B and among heterosexuals for CRF01_AE. 9,10 Subsequently, CRF01_AE appeared among MSM in 2005 11 and then experienced a rapid increase between 2006 and 2008, 2,12 and finally replaced subtype B as the most predominant strain in the MSM population after 2008. 13 Overall, cocirculation of CRF01_AE and subtype B among MSM within an identical high-risk population is likely to account for the generation of different unique recombinant forms of CRF01_AE and subtype B, as reported herein on two novel URFs among MSM.
In conclusion, the present study identified two novel URFs derived from subtype B and CRF01_AE among MSM in China. These new and unique recombinant forms of HIV-1 display a recombinant structure distinct from any other CRF or URF reported so far from China. As the cocirculation of multiple HIV-1 subtypes continues and the MSM population is prone to be infected by more than one virus, 14 it is likely that new recombinants of HIV-1 will continuously emerge in the MSM population, which highlights the importance of ongoing surveillance of HIV-1 subtypes and recombinant forms among MSM since this information has implications for HIV-1 vaccine development and new drug design and provides insight into the evolutionary history of HIV-1. Since partial genomic analysis is unable to identify the unique recombination, near full-length genomic analysis will be necessary for the investigation of the molecular epidemic among MSM.
Sequence Data
The near full-length genomic sequences of 08CYM015 and 08CYM047 have been submitted to GenBank with accession numbers JF340053 and JF340054, respectively.
Footnotes
Acknowledgments
We would like to thank both participants who contributed time, information, and blood samples for the study. This work was supported by Chinese National Grand Program on Key Infectious Disease Control and Prevention (2008ZX10001-015 and −002), Program for Outstanding Scientist in Shanghai (10XD1403500), and Leading Scientist in Medical Science [Shanghai Health Science and Education (2010-058)].
Author Disclosure Statement
No competing financial interests exist.