
Abstract
We have analyzed purine (R) and pyrimidine (Y) codon patterns in variable and constant regions of HIV-1 gp120 in seven patients infected with different HIV-1 subtypes and naive to antiretroviral therapy. We have calculated the relative frequency of each in-frame codon RNY, YNR, RNR, and YNY (N=any nucleotide) in variable and constant regions of gp120, in the sequence within indels and at indels' flanking sites. Our data show that hypervariable regions V1, V2, V4, and V5 are characterized by the presence of long stretches of RNY codons constituting the majority of the sequence portion within insertions/deletions. In full-length gp120 and within inserted/deleted fragments the number of AVT (V=A, C, G) codons did not exceed 50% of the total RNY codons. RNY strings in variable regions spanned up to 21 codons and were always in frame. In contrast, RNY strings in constant regions were mostly out of frame and their length was limited to five codons. The frequency of the codon RNY was found to be significantly higher in variable regions (p<0.0001; t-test), within indels, and at indels' flanking sites (p<0.0001; χ2 test). Analysis of the distribution of RNY strings equal to or longer than five codons in the full genome of HXB2 also shows that these sequences are mostly out of frame, unless they contain a potential N-glycosylation site or an asparagine. These data suggest that cryptic repeats of RNY may play a role in the genesis of multiple base insertions and deletions in hypervariable regions of gp120.
Introduction
T
In previous studies we have investigated within-patients genetic diversity in the fourth variable region of gp120. 14,15 Our data show that V4 is highly polymorphic within the same HIV-infected individual due to insertions/deletions (indels) of multiples of 3 base pairs, resulting in marked differences in sequence and length in V4 populations derived from the same clinical specimen. We have also shown that in V4, the nucleotide sequence of inserted/deleted fragments is often associated with the presence of elements of misalignment, such as palindromic sequences, long duplications, and repeated trinucleotides. 16 In our study, two hotspot motifs were identified in V4, a 15-mer and a 9-mer, namely SeqA and SeqB, which appeared to be conserved across subtypes and were often found to be duplicated in clones derived from the same individual. 16 On the basis of these findings we postulated that a mechanism of misalignment 17 –19 involving specific signal sequences, such as SeqA and SeqB and/or palindromes, could be responsible for at least some of the indels observed in V4. 16 However, although several more sequences spanning the entire gp120 molecule were screened, no elements similar to SeqA and SeqB that could be regarded as signal sequences triggering multiple base pair indels could be identified in variable regions other than V4 (E. De Crignis, unpublished results).
There is increasing evidence that both cruciform structures generated by the presence of palindromic sequences and simple repeating DNA sequences such as trinucleotides have mutagenic potential, due to their ability to adopt non-B DNA conformations. 20 –30 Mutagenic potential has been also associated with cryptic repeats of alternating purines and pyrimidines, such as the motif RRY (R=purine; Y=pyrimidine). 21,31 –34 Furthermore, triplets RNY (N=any nucleotide) have been shown to have a higher propensity to be separated by multiples of three nucleotides. 35 Codons characterized by cryptic RNY motifs such as AAT or AVT (V=A, C, or G) have been reported to appear with higher frequencies in the length-variable portions of gp120. 36,37 The ability of AVT codons to mediate size variation while being able to encode for glycosylation sites has been proposed to explain the high concentration of AVT motifs in V1, V2, and V4. 37
In this study we have investigated the distribution of the trinucleotide RNY in a fragment of Env spanning C1–C5 and encompassing all the variable domains of gp120. The distribution of the codons YNR, RNR, and YNY was also analyzed, in constant and variable regions of gp120, and within inserted/deleted fragments. Our aim was to assess whether cryptic patterns due to specific alternations of purines and pyrimidines could be recognized in variable domains of gp120 that could be regarded as hotspots associated with the presence of indels.
Materials and Methods
Patients
This study was performed on a cohort of seven patients screened randomly for a protocol approved by the institutional review board of the Centre Hospitalier Universitaire Vaudois (CHUV), Lausanne, Switzerland. Patients included in this study had a mean CD4+ T cell count 475±106/mm3.
Cloning and sequencing procedures
The RT-PCR and cloning procedures have been described elsewhere. 15 In all patients an Env fragment from HIV-1 plasma RNA spanning C1–C5 and corresponding to bp 6495→7757 of the HIV-1/LAV reference genome 38 was cloned and sequenced. In HXB2 this fragment is 1263 bp long. In total, 130 clones were analyzed in this study. As a positive control, bulk cDNA from culture supernatants of ACH-2, a T cell line chronically infected with HXB2, 39 was also amplified, cloned, and sequenced using the same conditions as those used for the clinical samples.
Sequence analysis
Gap-inclusive alignments of gp120 consensus sequences were created to align consensuses containing variable or constant regions of different length derived from the same patient. In gap-inclusive alignments, gaps were inserted between codons to conserve the reading frame of the protein.
Sequence numbering in gp120 is referenced to the HIV-1 HXB2 prototype using the HIV Sequence Locator Tool available at the HIV Sequence Database (
Sequence analyses were performed using Sequencher, Clustal X 1.83, and Bioedit software packages, together with the tools available at the HIV Sequence Database (
Analysis of codon content
Strings of consecutive RNY codons were visualized simultaneously in individual clones by the Motif Definitions key of the Sequencher package. For codon counts, the nucleotide sequence of each region of each clone was exported in Word, and divided manually in in-frame triplets. Each triplet was then translated into the corresponding codon (RNY, YNR, RNR, YNY) and the codons counted. The relative frequency of each codon type was calculated as the percent fraction of the length of the region expressed in total number of codons. When more than one variant was present for a given region, the percent value was calculated on the average number of codon types divided by the average number of total codons found in all the variants of that particular region.
Statistical analysis
The statistical significances of codon frequency in constant and variable regions of gp120 and of differences in the frequencies of strings of RNY ≥6 in viral genomes were calculated by unpaired two-tailed Student's t test; statistical significance of RNY distribution at indels flanking sites was assessed by the χ2 test. Statistical tests were performed using GraphPad Prism 5 and Excel.
Phylogenetic analysis
A phylogenetic neighbor-joining tree was constructed and used to verify that each consensus sequence segregated only with other sequences derived from the same patient. Consensus sequences aligned using GeneCutter (
Results
Seven patients were analyzed in this study, infected with three different HIV subtypes, namely B, C, and CRF02_AG. Phylogenetic relations among the different isolates and subtype assignments are shown in Fig. 1. In all patients major indels were identified in V1, V2, V4, and V5 derived from the same clinical specimen. As already reported in V4, 14,15 indels in other regions of gp120 also involved multiples of 3 base pairs. Neither frameshifts nor stop codons were ever observed within indels occurring in variable regions, although a 12-bp frameshifting indel due to a deletion of 13 bases, compensating a 1 base insert about 70 bases downstream, was observed in C2 of Patient 6 (data not shown). The data of intrahost length polymorphism obtained in the seven patients studied are summarized in Table 1. Values of clonal frequency calculated for each gp120 variant are issued from standard PCR reactions and are therefore relative to the total number of sequences identified in this study, which, in turn, are indicative of the variants most represented in plasma.
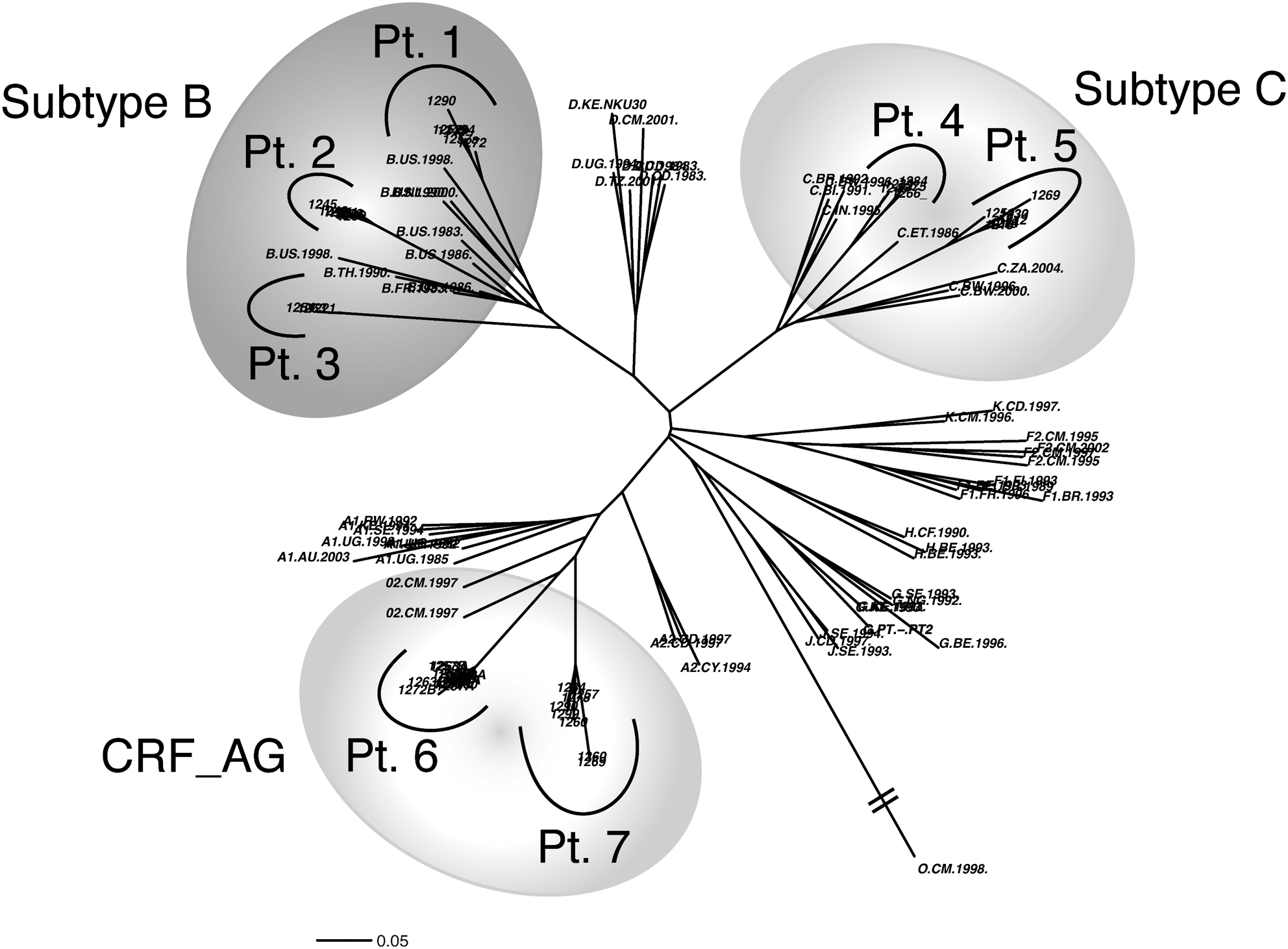
HIV sequence distances and subtype assignment. Maximum likelihood (ML) tree including all the consensus sequences derived from each patient and reference sequences from HIV-1 group M subtypes. The length-variant consensuses found for each patient cluster together and emerge from a unique branch of the tree, close to the reference sequences of the HIV subtype to which they are assigned. In each patient, consensus sequences were obtained by aligning clones with variable and constant regions of the same length. Reference sequences were obtained from the HIV sequence database at Los Alamos.
The length of individual regions is expressed in amino acids.
The length of whole gp120 molecules is expressed in base pairs. Letters indicate gp120 clones of equal length in which variable and constant regions differ in size and/or sequence.
Variable regions of gp120 are characterized by strings of consecutive RNY codons
In all patients, a pattern of alternating purines and pyrimidines was observed in variable domains of gp120 (Fig. 2). The pattern appears to be due to a sequence of two purines followed by a pyrimidine (RRY), with a few interruptions due to point mutations occurring mostly in the second position of the codon. In V1 of Patient 7 (clone 1278), two strings can be recognized, formed by five consecutive RRY. However, if we ignore the second nucleotide of the codon and consider only the purine and the pyrimidine in the first and third position, the pattern becomes RNY, represented by a string of 17 consecutive units. An analogous situation can be seen in V5 of Patient 5 (clone 1254), in which a string of 4 RRY becomes a sequence of 10 consecutive RNY (Fig. 2).
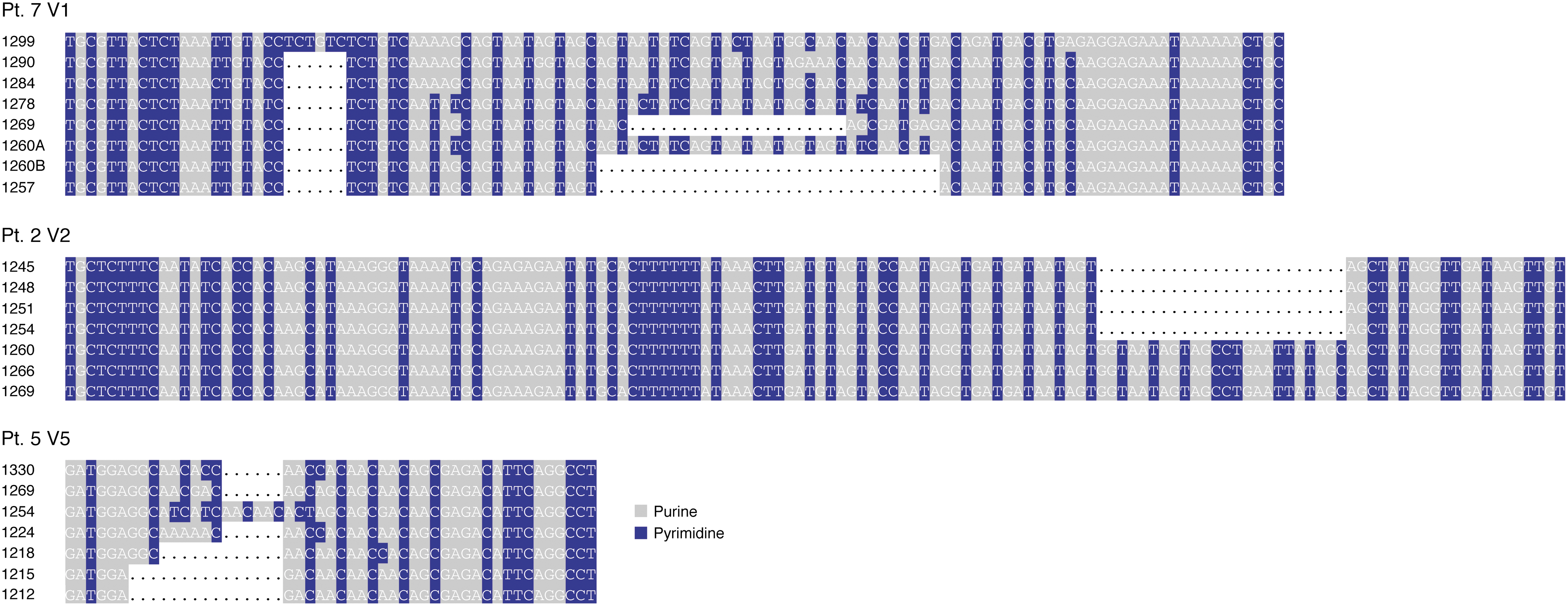
Association between patterns of alternating purines and pyrimidines and indels in gp120. Gap-inclusive alignments of V1, V2, and V5 in three patients chronically infected with HIV-1 and naive to antiretroviral therapy. Purines (A, G) are shaded in gray and pyrimidines (C, T) in blue. Missing sequences within indels are indicated by dots. Gp120 clones are indicated by the total length of the amplified fragment expressed in base pairs.
Stretches spanning four or more consecutive RNY are found preferentially in hypervariable regions of gp120
Figure 3 shows a schematic representation of the distribution of individual RNY codons in gp120 of HXB2. Single RNY triplets are homogeneously distributed across the whole molecule and can be consecutive, overlapping, in frame, and/or out of frame. However, if we increase the string size by progressive addition of individual codons, we can observe a gradual disappearing of multiple RNY motifs from most of gp120, with preferential localization in hypervariable regions.
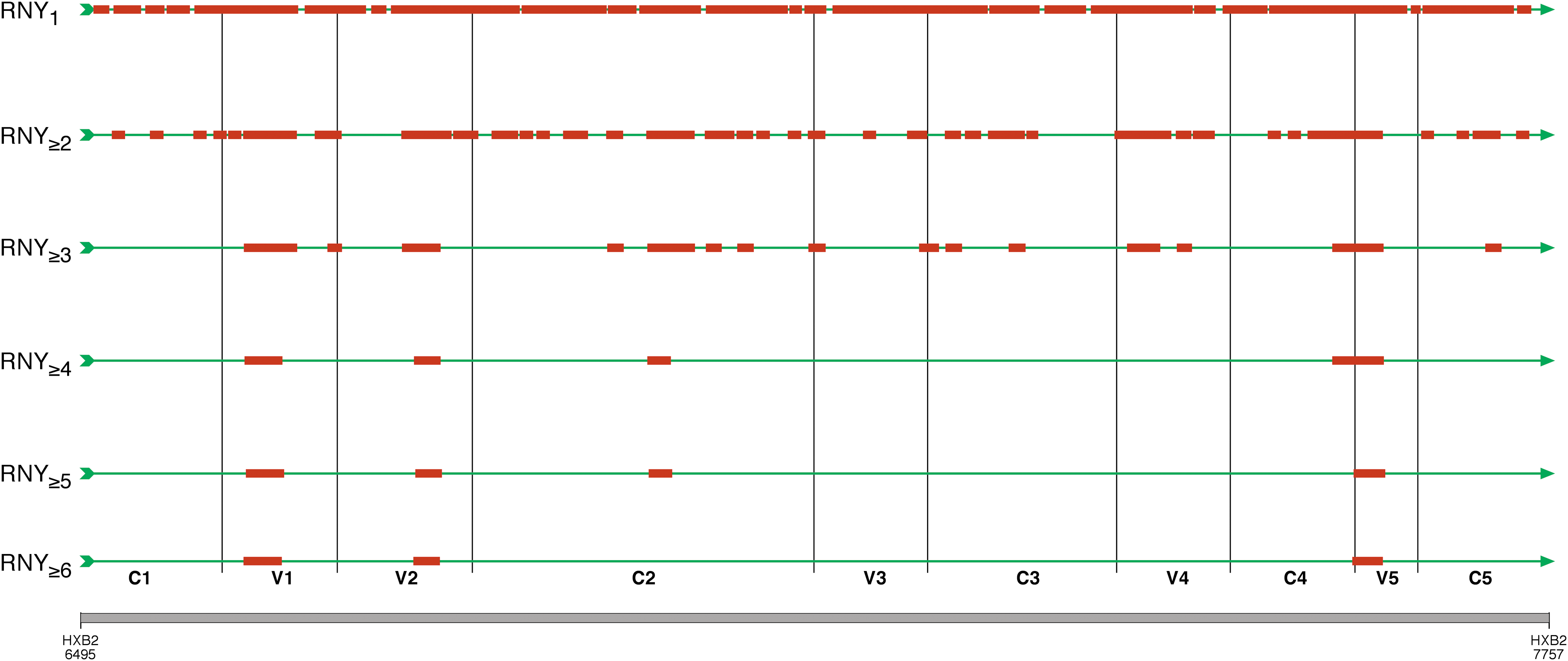
Localization of RNY strings of increasing length in gp120 of HXB2 (bp 6495–7757 of HXB2). In the first line, the distribution of single RNY codons across gp120 is shown. At each of the following lines, the length of the RNY repeat is increased by 1 codon. The second line represents the distribution of RNY repeats of at least 2 codons and the third line the distribution of strings of at least 3 codons. In the last line, the distribution of RNY strings made of at least 6 codons is shown. RNY strings are shown in red. Green arrows indicate the gp120 fragment.
A similar situation can be observed in patients. Table 2 shows the distribution of strings equal to or longer than four codons (RNY ≥4) in 46 clones derived from the seven patients studied. All the clones selected carried a unique RNY pattern and were therefore all different from one another. Each string is represented by a number expressing the number of consecutive RNY codons constituting the string itself. For example, two strings are present in V1 of clone 3–1263, spanning 4 and 5 codons, respectively. Within the same patient, strings present in the same column correspond to the same sequence and therefore they can be aligned. No RNY ≥4 were observed in the portion of C1 amplified in this study and in V3 (data not shown). Stretches spanning up to 21 RNY were observed in V1 and up to 10 in V2. Stretches up to 10 or 11 RNY codons were also observed in V4 and V5. C2 was characterized by the presence of small strings of in-frame RNY spanning 4 codons, and by one or two strings spanning 4 or 5 codons that were out of frame. Out-of-frame strings ≤5 codons were also present in C3 and C4. No strings of out-of-frame RNY longer than 5 trinucleotides were observed.
In-frame strings of RNY≥4 are shown. Bold characters indicate strings of RNY≥4 codons that are out of frame. Only regions in which strings of RNY≥4 are present are shown.
Numbers in each column refer to how many RNY codons are present in each string.
The majority of codons with the motif RNY are of the non-AVT type
Codons AVT and AAT have been reported to coincide with length-variable regions of gp120.
36,37
We calculated the fraction of RNY constituted by the motif AVT, which also includes the motif AAT. RNY non-AVT and AVT codons were counted in full-length gp120 in the 46 unique clones listed in Table 2. In all, 4836 codons were counted, divided into 2214 AVT and 2622 RNY non-AVT, which corresponded to 54.2% of the total. As shown in Table 3, in individual patients, the cumulative percent of codons RNY non-AVT in these regions ranged from 47.5% (Patient 7) to 60.1% (Patient 2). Next we counted only codons RNY non-AVT and AVT within inserted/deleted fragments in regions V1, V2, V4, and V5. This time 345 codons were, counted, 186 of which were RNY non-AVT, corresponding to 53.9% of the total, although with some more variation among individual patients (82.5% in Patient 5, 29.4% in Patient 4) (Table 3). Strings of four or more AVT codons were found only in 9 of the 46 clones analyzed (Supplementary Table S1; Supplementary Data are available online at
Regions V1, V2, V4, and V5.
The frequency of individual RNY codons is significantly higher in variable than in constant regions of gp120
The relative frequency of each of the four codons RNY, YNR, RNR, and YNY in the various regions of gp120 was assessed by counting each codon in each region of each clone. Only codons that were in frame were counted. The percent values of the frequencies obtained in all variable regions for each codon were plotted against the percent values of the frequencies obtained in all constant regions for the same codon. A t-test performed by the GraphPad Prism 5 package shows that the frequency of RNY codons is significantly higher (p<0.0001) in variable regions than in constant ones (Fig. 4). Conversely, the frequency of the codon YNR is significantly lower in variable regions (p<0.0001). The frequency of codon RNR is also higher in constant regions, although with a higher p value (p=0.0178), whereas no differences were observed in the frequency of codon YNY. Analysis of codon distribution in the sole region V3 shows no significant differences in the frequency of codons RNY, YNR, and YNY. In contrast, the relative frequency of codon RNR was significantly higher (p<0.0001) in this region (data not shown).
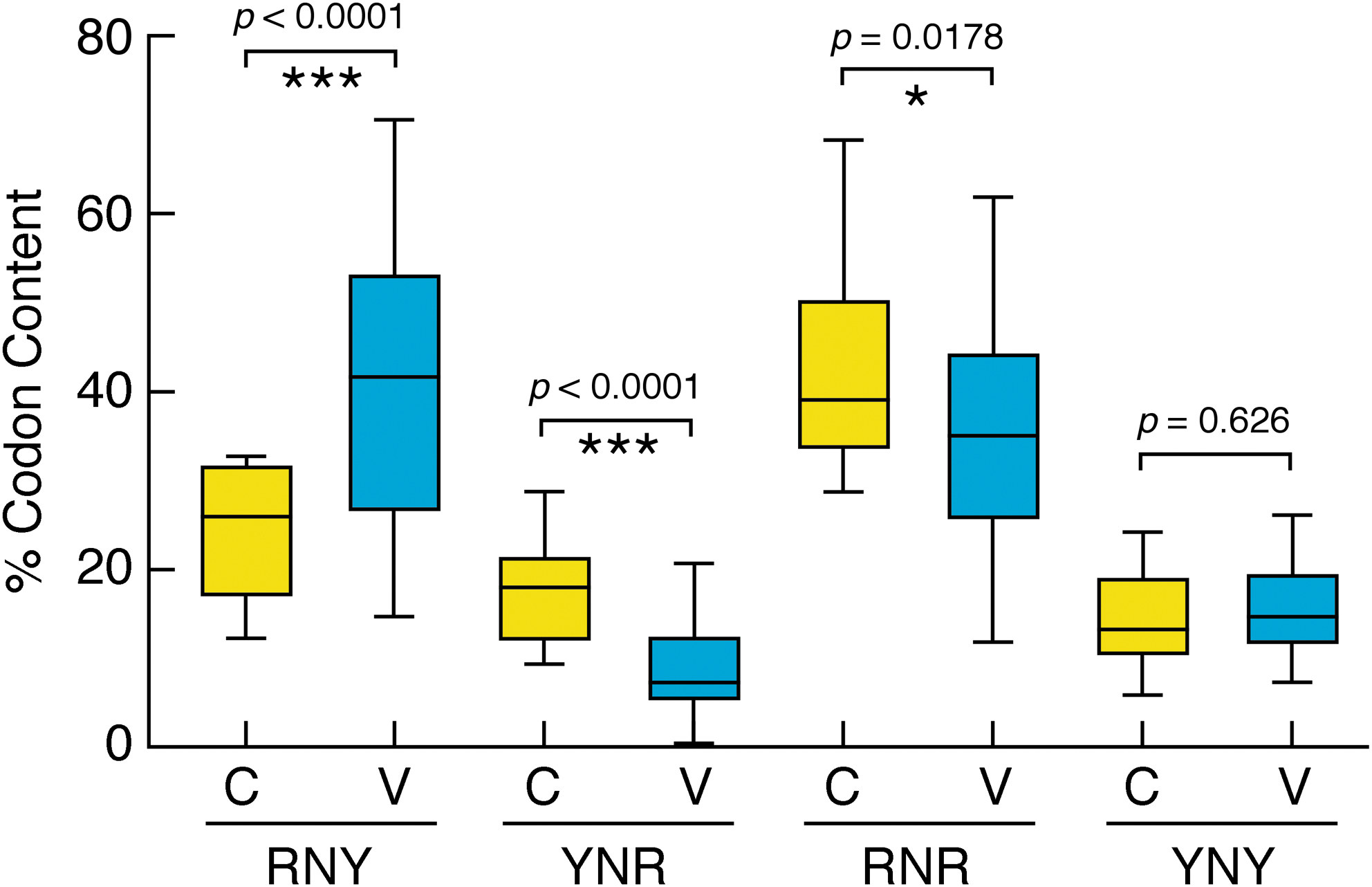
Whisker plot showing the differential distribution of codons RNY, YNR, RNR, and YNY in variable (V) and constant (C) regions of gp120. Codons RNY are significantly more frequent in V than in C regions (p=0.0001; unpaired t-test). Yellow boxes indicate the percent frequency of each codon in constant regions. The relative frequency of codons in variable regions is indicated by blue boxes.
RNY codons are preferentially found within inserted/deleted sequences and at indels' flanking sites
The codon content of the sequences in inserted/deleted fragments and in indels' flanking sites was also analyzed. In each patient, individual V regions carrying indels were aligned and in each clone the codons within indels were counted. In total, 83 indels were analyzed, distributed as follows: 29 in V1, 19 in V2, 25 in V4, and 10 in V5. As shown in Fig. 5A, most of the sequences within indels consist of RNY codons (≥50%). Of the other codons, RNR accounts for about 30%, YNY for 20%, and YNR for less than 10%. The codons at the right and left side of each indel were also counted (Fig. 5B). RNY trinucleotides were found to account for 76% of the total left and 64% of the total right flanking codons (p<0.0001; χ2 test).
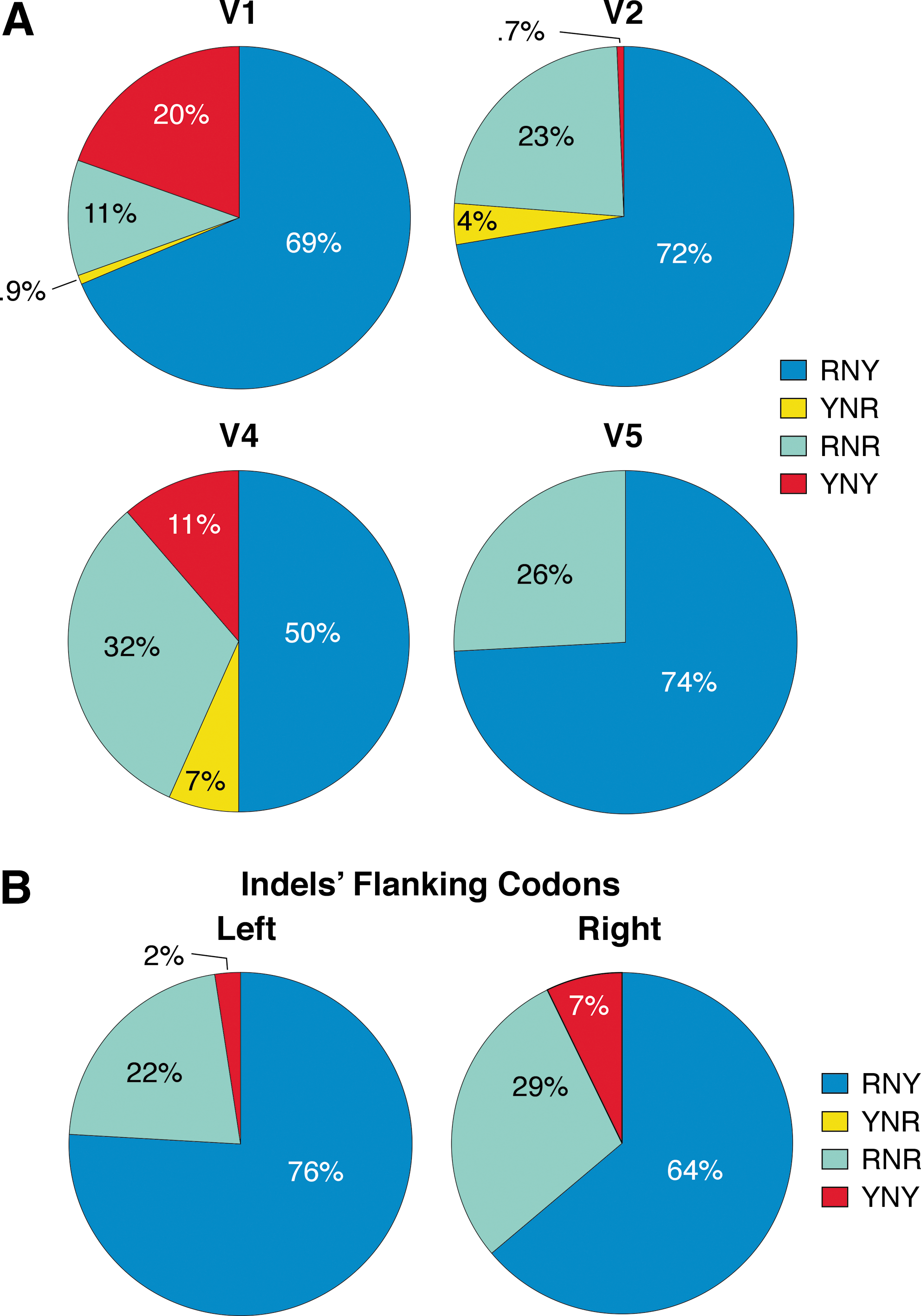
Prevalence of RNY codons within indels and at indels' flanking sites.
In the genome of HXB2, strings of RNY spanning four or more codons are mostly out-of-frame
Besides those described in gp120, a total of 23 additional RNY ≥4 were found throughout the entire HXB2 genome (Table 4). Sixteen were out of frame. Of the seven that were in frame, four contained an asparagine (N) and one contained a potential N-glycosylation (PNG) site. Only two RNY strings that were in frame and contained neither an asparagine nor a PNG site were found. One of them, however, in Gag p24, contained the sequence AGT ACC [serine (S)–threonine (T)] (HXB2 bp 1504–1515), which constitutes two thirds of the PNG site consensus (NXS/T). A string spanning 17 RNY was also found in Vpu (HXB2 bp 6073–6123), in a region known to be polymorphic. 40
In frame.
In frame containing an asparagine residue.
In frame containing a PNG.
Each symbol refers to an individual string (e.g., a,b means three strings, one in frame involving an asparagine, one in frame, and one out of frame).
The frequency of RNY strings of six or more codons is higher in HIV-1 than in other retroviruses
To assess how widespread repetitive RNY codons are in viruses, we counted RNY strings in the full genome of 55 viral species derived from GenBank and from the HIV database, which included lentiviruses, other retroviruses, and DNA and RNA viruses (Table 5). Given the fact that strings of RNY spanning 4 or 5 codons are quite frequent on a genome scale, and that indels in variable regions are associated with strings made of at least 6 codons, we decided to count only RNY strings consisting of at least 6 repeats. The results of this analysis are shown in Table 5. Numbers in each column represent how many strings of that particular length (expressed in number of RNY codons) are present in each genome. We found that the number of consecutive RNY spanned up to 17 repeats, except in the case of Molluscum contagiosum virus (MCV), a double-stranded (ds) DNA virus in which strings spanning up to 21 and 34 consecutive RNY were observed. The average frequency of each string length (number of RNY n /kb) calculated in each group of viruses was compared to the average frequency of the same string length calculated in all the genomes analyzed. The frequency of RNY strings longer than 6 codons was found to be slightly higher in HIV-1 than in HIV-2, SIV, and other lentiviruses and similar to the one observed in dsDNA viruses (p=0.01; t-test).
Discussion
In this study we have analyzed the nucleotide sequence of a fragment of gp120 spanning C1–C5 in HIV plasma RNA derived from seven patients infected with different HIV subtypes and naive to therapy. Our aim was to assess whether a correlation could be established between the presence of cryptic patterns of alternating purines and pyrimidines and the occurrence of multiple base insertions and deletions in variable regions of gp120. Specifically, we have mapped the distribution of strings of RNY equal to or longer than four codons in gp120 derived from patients and from HXB2. Furthermore, we have analyzed the frequency of each individual codon RNY, YNR, RNR, and YNY in constant and variable regions of gp120, in the sequence within indels, and at indels' flanking sites. In all patients, multiple variants of each region, mostly V1, V2, V4, and V5, but in some cases also C2 and C3, were observed to coexist simultaneously.
Different variants were characterized by the presence of insertions and deletions spanning several bases, which were multiples of 3 base pairs, with no generation of frameshifts and stop codons, with the exception of a frameshift deletion in C2 of one patient (Patient 6). These observations are consistent with the patterns of multiple base pair insertions and deletions during early infection described by Wood and co-workers, 41 with in-frame indels, presumably functional, occurring only in hypervariable loops, and out-of-frame ones distributed throughout Env.
We show that the majority of the sequence fraction that is inserted/deleted in V1, V2, V4, and V5 consists of long stretches of in-frame, repeating RNY units. A major feature of interest is the cryptic nature of the sequence within indels: in fact, if these fragments are compared only on the basis of their sequences, they share only partial similarity, and do not appear to be consistent among different gp120 regions and viral isolates. However, once these fragments are translated into sequences of purines and pyrimidines, the overwhelming presence of RNY codons, and consequently the repetitive nature of the sequences involved in indels, becomes evident. RNY considerably expand the range of triplets that can be involved in N-glycosylation.
In previous studies, 37 codons with the motif AVT have been reported to be present in high percentage in V1/V2, encoding for asparagine (AAT), serine (AGT), and threonine (ACT). AVT, however, represents less than 50% of the total population of RNY in HIV-1 gp120. Furthermore, the presence of clusters of AVT in all variable regions of gp120, including V5, is still controversial. 37 However, if AVT is analyzed from an RNY standpoint, it appears in higher percentage also in V4 and V5, encoding not only for asparagine (AAT, AAC), serine (AGT, AGC), and threonine (ACT, ACC), but also for glycine (GGT, GGC), alanine (GCT, GCC), valine (GTT, GTC), isoleucine (ATT, ATC), and aspartic acid (GAT, GAC). In the length-variable portion of gp120, codons AVT and RNY non-AVT are interspersed. Strings made by either one of these two motifs alone are very short and distributed unevenly among the different regions, masking the recurrent RNY pattern. As a consequence, focusing on the sole motif AVT did not make it possible to realize that inserted/deleted regions are constituted almost exclusively by long stretches of RNY consecutive codons.
In hypervariable regions V1, V2, V4, and V5, RNY strings are always in frame. RNY strings in constant regions of gp120 and across the rest of the genome of HXB2 are mostly out of frame, unless they contain a PNG site or an asparagine. Asparagine is the amino acid that is central to the PNG site, a tripeptide whose consensus sequence is N-X-[S or T], where X can be any amino acid (except proline) followed by serine (S) and threonine (T). 42,43
Changes in the number and patterns of potential N-glycosylation sites have been shown to increase the ability of the virus to escape the host immune system 44 –48 and hypervariable regions of gp120 have been shown to function as hotspots for PNG site rearrangements. 36,49 Based on these data, it appears that mechanisms able to increase the probability of reshuffling of PNG sites in variable regions of gp120 are likely to be favored by selection, since they would also lead to an increase of the virus escape potential. Thus, selective pressure would act toward the presence of several consecutive RNY codons in regions that can tolerate change (hypervariable domains); in addition, selective pressure would favor the occurrence of the reading frame “RNY” (in frame), as opposed to “NYR” and “YRN” (out of frame) in those regions, since it would be the only one leading to PNG site pattern rearrangements without disruption of the PNG site.
Thus, the finding of in-frame RNY strings mostly in variable domains would be the result of positive selection in regions in which PNG site reshuffling may confer selective advantage. It should be also pointed out that PNG site reshuffling is not limited to AAT or AVT codons. In fact, the codons of the PNG site can have both the motifs AVT and RNY non-AVT. The codons AAT (asparagine), AGT (serine), and ACT (threonine) belong to the AVT subgroup, whereas AAC (asparagine), AGC (serine), and ACC (threonine) are RNY non-AVT. In addition, in inserted/deleted fragments, RNY non-AVT and AVT codons are equally represented, with a slight prevalence of codons RNY non-AVT.
We analyzed the genomic distribution of strings of RNY ≥6 in 55 additional viral genomes including lentiviruses, retroviruses, and DNA and RNA viruses. To our surprise, the frequency of RNY repeats observed in HIV-1 was higher than the frequency observed in all the other groups of viruses, with the exception of dsDNA viruses. We searched the literature to see whether events of length polymorphism and indels similar to those observed in HIV-1 had been reported also in dsDNA viruses. However, since most of the sequencing data available for these viruses come from the analysis of just a few laboratory strains or clinical isolates passaged in vitro, very little information is available about mutations in wild-type isolates. In the case of herpes simplex virus (HSV-1), for example, until very recently, only one wild-type genome sequence was available, which was completed over 20 years ago. 50 –52
Mutations occurring during retroviral replication have been unanimously considered as the primary cause of HIV-1 diversification. 53 –56 Although it is widely accepted that the low fidelity of RT is the major cause of single base pair mutations, the genesis of multiple base pair insertions and deletions has yet to be elucidated. Abram and co-workers 57 have investigated the role of polymerases (RT, DNA-dependent RNA polymerase during RNA transcription and of host DNA-dependent DNA polymerase) in mutations generated during a single cycle of HIV-1 replication. Although some of the mechanisms underlying the mutations remain undefined, their data clearly indicate that different mutations are due to different mechanisms.
During the past two decades, expansions of triplet repeats have been involved in a number of genetic diseases, such as fragile X syndrome, myotonic dystrophy, and Huntington's disease. 58 Our data show that the length-variable portion of gp120 is constituted almost exclusively by cryptic repeats of the triplet RNY, and that multiple base pair insertions and deletions are due to variations in the number of the RNY codons present within the inserted/deleted fragments. These results may suggest that multiple base pair indels in gp120 (and possibly in other regions of the HIV-1 genome) may be generated by a mechanism of triplet repeat expansion. The onset of new RNY codons in gp120 would be a random process, due to single base pair mutations caused by the low fidelity of RT and regulated by selection. Since accumulation of RNY codons increases the probability of occurrence of insertions and deletions, viral genomes carrying RNY clusters in regions required to remain “constant” would be selected against. In contrast, genomes characterized by RNY clusters in regions prone to change would be favored by selection. This would explain the presence of high concentrations of RNY mostly in variable regions of gp120. Further studies are needed to define the role of RNY repeats in the generation of indels and to elucidate the mechanism(s) underlying these mutations.
Nucleotide Sequence Accession Numbers
The GenBank accession numbers for the sequences generated in this study are HQ322131 to HQ322260 (
Footnotes
Acknowledgments
We would like to acknowledge the patients and all the people in the lab who contributed to the realization of this study. Special thanks to John Weddle for the artwork and to Matthieu Perreau for his help with Graphpad Prism.
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.