
Abstract
To characterize phylogenetic relatedness of plasma HIV-1 RNA subtype C env gp120 viral variants capable of establishing an infection following heterosexual and subsequent vertical transmission events a 650-base pair fragment within the C2-V5 subregion was sequenced from four HIV-1-infected families each consisting of biological parent(s), index children (first), and subsequent (second) siblings. None of the family members had received antiretroviral therapy at the time of sample collection. Sequence alignment and analysis were done using Gene Doc, Clustal X, and MEGA software programs. Second siblings' sequences were homogeneous and clustered in a single branch while first siblings' sequences were more heterogeneous, clustering in separate branches, suggestive of more than one donor variants responsible for the infection or evolution from founder variant(s) could have occurred. While the directionality for heterosexual transmission could not be determined, homogeneous viral variants were a unique characteristic of maternal variants as opposed to the more heterogeneous paternal variants. Analysis of families' sequences demonstrated a localized expansion of the subtype C infection. We demonstrated that families' sequences clustered quite closely with other regional HIV-1 subtype C sequences supported by a bootstrap value of 86%, confirming the difficulty of classifying subtype C sequences on a geographic basis. Data are indicative of several mechanisms that may be involved in both vertical and heterosexual transmission. Larger studies are warranted to address the caveats of this study and build on the strengths. Our study could be the beginning of family-based HIV-1 intervention research in Zimbabwe.
Introduction
S
Single or multivirus transmission event(s) can initiate HIV-1 infections. 9 –11 Despite the fact that the transmitter harbors heterogeneous variants, newly transmitted HIV-1 gp120 env sequences in the recipient show relative uniformity until the immune responses drive the founder virus to diversify into a quasispecies, closely related viral swarms. 3,12 –15 HIV-1 subtype B studies have shown a linear increase in env diversity during the first few years of infection, which tends to stabilize or even decrease at some point but often becomes homogeneous once more as the immune system wanes. 16 –18 The founder variants have been shown to reappear as the dominant quasispecies in plasma later during infection. 19 Diversity of recent heterosexually transmitted variants has been shown to be greater in women than men. 20 Studies have shown that the use of hormonal contraceptives and sexually transmitted infections increase the likelihood of acquiring heterogeneous variants from a single donor. 21 –23 Acquisition of homogeneous viral quasispecies is a unique and consistent feature of perinatal HIV-1 transmission, suggesting the presence of selective host pressures. 24 –26 However, some studies have reported infants with exclusive heterogeneous viral populations, 27,28 while other studies suggest multiple mechanisms with different proportions of both homogeneous and heterogeneous viral populations. 29 –32 At least for HIV-1 subtype A, homogeneous viral populations have been observed in 50% of vertically infected infants. 33
The current knowledge on HIV-1 transmission is biased toward homosexually or parenterally acquired subtype B and is confined to Europe and America at the expense of the most widespread subtype C. Phylogenetic assessments of the HIV-1 subtype C env gp120 region during both acute and chronic infections are limited in Zimbabwe, particularly, following concurrent heterosexual and vertical transmission events. Four HIV-1-infected families provided an opportunity to characterize phylogenetic relatedness of the virus variants capable of establishing an infection following heterosexual and subsequent vertical transmission events by genotyping a 650-base pair fragment of the HIV-1 subtype C env gp120 C2-V5 subregion.
Materials and Methods
Study population and procedures
Four HIV-1-infected families labeled 205, 366, 375, and 567 consisting of biological parent(s)-infected siblings, an index child (older), and an index child's sibling (younger) constituted the study population. The unit of analysis was a family and these four were willing to participate in the phylogenetic study. The index child in this study was defined as the first child to be recruited into our study. Two families comprised both parents and a respective biological index child. The other two families had parent(s) and two subsequent biological children, the first and second siblings. In family 567 the father figure was missing as he was working in another regional country. Each member of the four families was HIV-1 infected and none had received antiretroviral therapy at the time of sample collection. We hereby describe a unique HIV-1 transmission clusters of four families for which the time and directionality of vertical transmission were known but were unknown for heterosexual transmission.
Consent was obtained from the four pregnant mothers of each family participating in the national PMTCT program in the periurban Harare mother and child clinic who were known to be HIV-1 positive at 36 weeks gestations. Spouses also consented to participate in the family phylogenetic study. Similar recruitment and procedures were followed as previously described for the mothers and infants. 34 Despite being encouraged to exclusively breastfeed during the first 6 months of life, all the infants were exposed to breast milk for at least 9 months. First siblings' plasma samples were collected at 60±10 months of age as there were insufficient sample volumes from their respective first HIV-positive samples. The first available HIV-1-positive sample was genotyped for the second sibling at about 15±3 months; for details see Table 1. Sexually transmitted infection (STI) screening, nucleic acid extraction, polymerase chain reaction (PCR) amplification, cloning, and DNA sequencing methods for the HIV-1 env gp120 C2-V5 region were done as previously described and so was HIV-1 subtype determination. 35 Briefly, the primary PCR amplified an approximately 800-base pair (bp) fragment spanning the C2 and V5 region of the envelope, positions 6948–7537 in the HIV-HXB2 genome while the secondary PCR amplified an approximately 650-bp env gene fragment.
F, father; M, mother; 1st, first siblings; 2nd, second siblings; gender symbols denoting male ♂ and female ♀.
DNA sequence analysis
Phylogenetic and molecular evolutionary analyses are pivotal in the clarification of transmission patterns of HIV. To visualize the extent of the genetic relatedness of the HIV-1 env gp120 C2-V5 region among members of the four families following heterosexual and vertical transmission env amino acid sequences were analyzed including the construction of a phylogenetic tree using MEGA 5.0 software. 36 Using the Clustal X program positions with gaps were excluded before tree building, consequently DNA sequences had an equal length before alignment. The most popular test for tree reliability, bootstrap, was used. The bootstrap value is a percentage of how often each branch is present in exactly the same topology in all the resampled trees. A bootstrap cut-off value of >70% signified at least a 95% probability that the topology of a branch is real. 37 An HIV-1 group O sequence obtained from the Los Alamos national database of HIV Sequence Compendium 2009 was used as the outgroup for rooting each of the families' trees. 38 Families' sequences were compared with other HIV-1 group M subtype reference sequences retrieved from the same database including other subtype C sequences from different countries within the SSA and other geographic regions such as Argentina, China, and India.
Genetic distance between two HIV-1 sequences is the count of the number of differences arising due to mutations and genetic drift resulting in genetic diversity. Differences were computed using Tamura and Tamura-Nei distribution-based distances also in MEGA. 36 Diversity, which is a measure of genetic variation at a given time, was calculated by measuring nucleotide diversity (π) implemented in MEGA 5. Homogeneity or heterogeneity of the C2-V5 regions in the viral clones of fathers, mothers, and first and second siblings was evaluated by comparing the number of unique DNA sequences among multiple clones per family member. Comparison of the genetic diversity and maximum genetic distance between families and different groups of family members, fathers, mothers, index children, or index children siblings was done. An arbitrary cut-off value of less than 1% was used to define viral homogeneity. 20
Results
Characteristics of the four families
Three of the four mothers were in a monogamous marriage except for mother 375 who was in a polygamous relationship. All parents had at least 7 years in school and were of low economic status. Mothers were generally 5 years younger than their spouses. Table 1 summarizes the demography of family members.
HIV infections
Parents of the four families were HIV-1 positive but did not know when and how they got infected. Parents' mode of HIV-1 acquisition was most likely heterosexually as none mentioned any history of blood transfusion, drug abuse, or homosexuality except for one mother, 366, who had a history of blood transfusion. The index children of families 366, 375, and 567 and second siblings of families 205 and 567 were HIV-1 DNA PCR negative at delivery and 6 weeks postpartum but later became infected through breastfeeding. Index child 205 was HIV-1 DNA PCR negative at delivery but was not at the 6 week visit, hence the exact time of infection could not be established.
Mothers' reproductive health and single-dose nevirapine (SdNVP) prophylaxis
All mothers had spontaneous vaginal deliveries. One life-time sexual partner was generally reported except for mother 375 who reported two. Due to religious beliefs mother 205 never used any method of contraception and moreover she refused to take SdNVP for herself and for both her infants. Otherwise the other mothers were on oral contraceptives and received SdNVP for themselves and their respective infants. All mothers had negative results for the RPR syphilis test but two of the four mothers were HSV-2 positive at enrollment. 39 Mothers 375 and 567 reported itchy genitals but no discharges were observed upon examination.
Subtypes and phylogenetic analysis
A total of 64 sequences from four family members were cloned of which a total of 35 unique clones were analyzed. On average four (three–five) clonal nucleotide sequences were determined for the env C2-V5 gp120 env region (650 bp) from each infected family member. Phylogenetic analysis of env amino acid sequences showed that HIV-1 subtype C had infected each one of the four families' members. The neighbor-joining phylogenetic tree showed that sequences were genetically linked and formed interfamilial clusters of HIV as shown in Fig. 1. Clusters were clearly distinguished with high bootstrap values suggestive of infections of monophyletic origin or a localized expansion of the subtype C epidemic at least among these four families. Our families env C2-V5 sequences clustered with other HIV-1 subtype C from South Africa, Malawi, Botswana, Tanzania, India, China, Argentina, and also from other previous Zimbabwean subtype C studies as evidenced by a bootstrap value of 86% (see Fig. 1). Sequences from families 205, 375, and 567 clustered more closely with sequences from South Africa, Malawi, and Botswana.
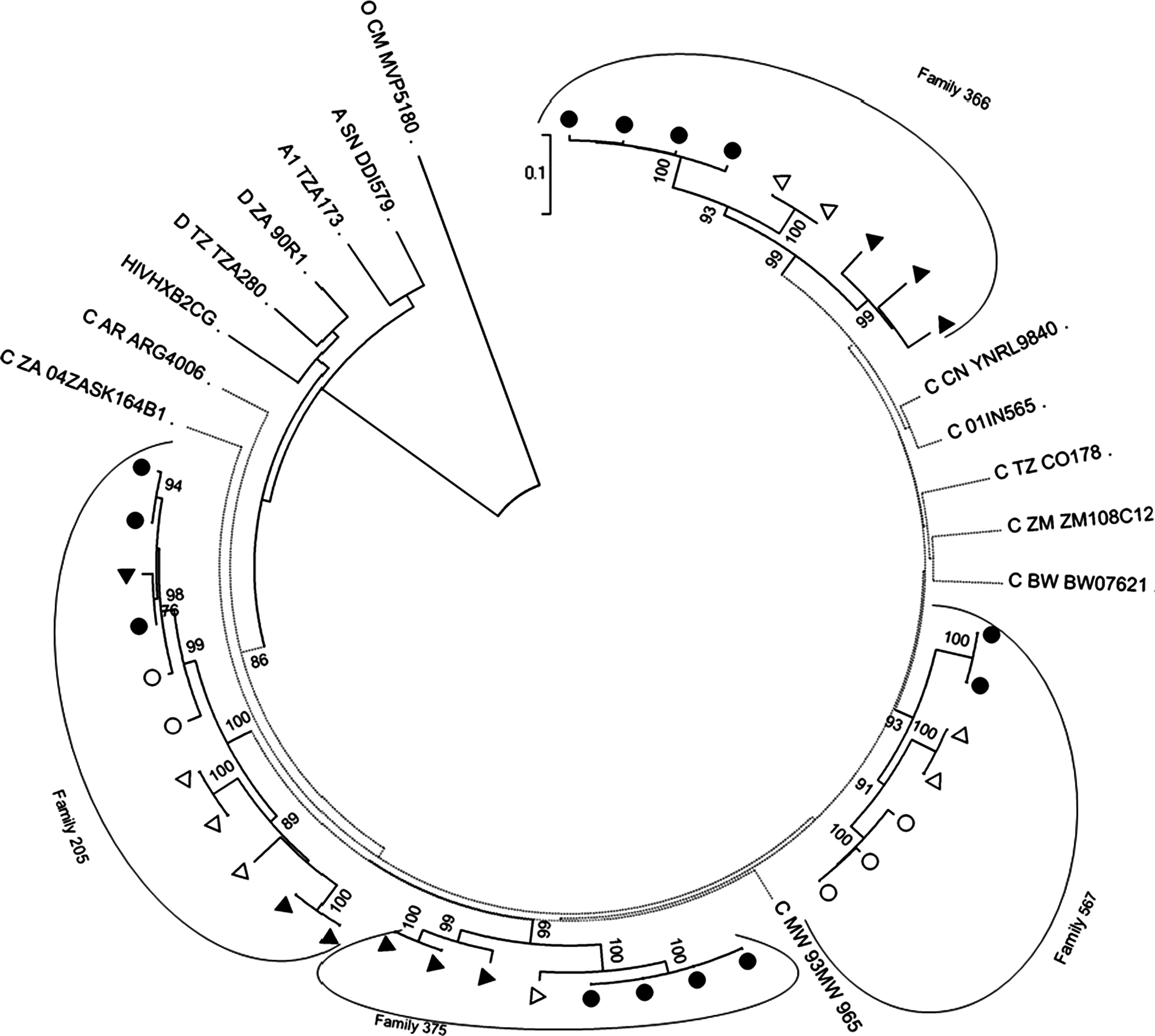
Phylogenetic relationships between families' nucleotide sequences and other subtype C from different geographic regions. The first letter represents the subtype followed by the name of the country: AR, Argentina; BW, Botswana; ZA, South Africa; TZ, Tanzania; IN, India; ZM, Zimbabwe; CN, China. Shaded triangles and circles represent fathers' and mothers' sequences, respectively, while open triangles and circles represent first and second siblings sequences, respectively. Bootstrap values are expressed as percentages per 1000 replicates and only proportions of ≥70% are shown.
Genetic distances
Although all the families sequences turned out to be subtype C with respect to the env gene, the interfamilies mean genetic distances varied being furthest apart between families 205 and 366 (20%) while families 205 and 567 showed the least genetic distances of 16.5% (see Table 2). The mean pairwise genetic distance was higher among the fathers, mean 18.13% (0.00–28.38), being highest between fathers of families 205 and 366. Mothers' percentage mean genetic distances were significantly lower, 17.21% (0.00–24.28), and the longest distance was between mothers of families 366 and 375. The mean genetic distance was statistically higher among the adult population, 16.5% relative to that of children, 11.5%. The intergroup mean genetic distance between fathers and first siblings was 17.7% (see Table 3).
F, fathers; M, mothers; 1st, first siblings; 2nd, second siblings.
Heterosexual transmission
Transmission events were epidemiologically linked, supported by high bootstrap values, suggestive of predominantly monogamous relationships. Paternal sequences showed the highest average number of unique isolates by nucleotide sequence. Father 205 had the most heterogeneity followed by 366 and then 375. Comparison of all viral populations revealed that fathers' sequences exhibited the most heterogeneous viral quasispecies, which were observed to cluster in several separate branches suggestive of multiple variants. However, mothers showed a consistent pattern of limited viral diversity. Consequently both single and multiple transmission events may have occurred in these close contacts. Since the direction of transmission was not known we could not show how diversity changed with heterosexual transmission.
Vertical transmission
Mothers and second siblings had limited heterogeneity, indicative of a relatively recent infection or suggestive of vertical transmission of a single or very few closely related maternal variants. However, a more heterogeneous virus population was generally observed for first siblings of families 205, 375, and 366, demonstrated by the intermingling of their sequences with the parental ones. Viral sequences were distributed into several branches suggesting multiple distinct lineages probably as a result of evolution away from maternal viral sequences through immune selection pressures.
Phylogenetic analysis of family 205 sequences
Figure 2a represents the neighbor-joining phylogenetic tree for family 205 family member's sequences intermingled with each other. Despite the limited number of clones per family member's sample, genetic heterogeneity was detected in the father's sequences, which intermingled with sequences from the mother and both the two children. The less prevalent paternal strain clustered in a single branch of the tree supported with a 98% bootstrap value with both the maternal and second sibling variants. The most prevalent strain of the first sibling appeared on its own branch supported with the lowest bootstrap value of 79%. The topology of the tree reflects the vertical transmission of at least two maternal variants that diverge over time as seen in the first sibling's sequences. It is worthwhile to note that in this family there was no SdNVP prophylaxis selection pressure.
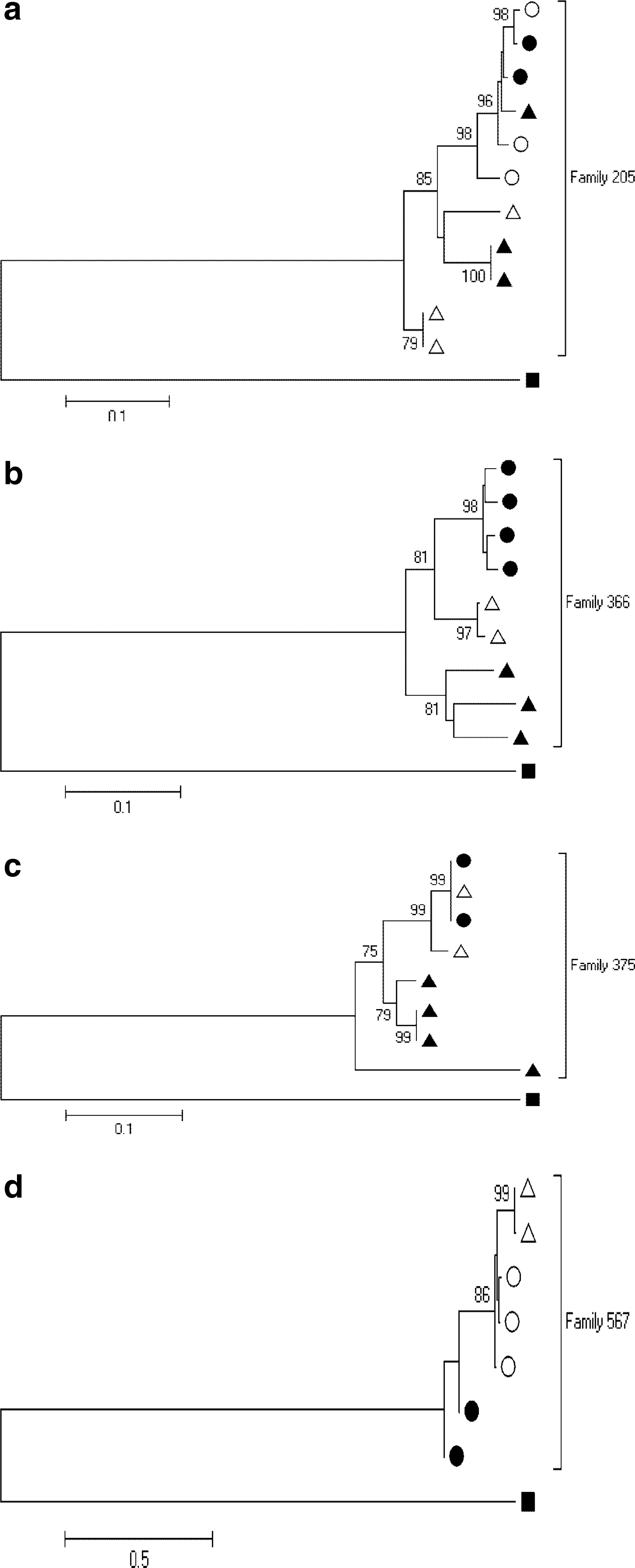
Rooted neighbor-joining trees of HIV-1 env (C2-V5) amino acid sequences for four family members
Phylogenetic analysis of family 366 sequences
Figure 2b represents the neighbor-joining phylogenetic tree for family 366. The high bootstrap values of >95% indicated the sequences are closely related and monophyletic. In contrast to family 205 sequences, homogeneous infections were presumed based on the level of genetic diversity. The child sequences were in between the parent sequences. All sequences of mothers and child clustered tightly on one branch with paternal sequences on the other, but all sequences were supported with a bootstrap value of 81%. In this case the child could have been infected with a single maternal variant, but because of the time factor in between, the variant could also have evolved under host immunological selective pressure.
Phylogenetic analysis of family 375 sequences
Like family 366, paternal sequences clustered tightly on one branch while the mother–child sequence also clustered tightly on another branch with a bootstrap value of 99%, indicative of a relatively more recent infection, as shown in Fig. 2c. The overall family tree is supported by a high bootstrap value. As with family 205, the child sequences were also intermingled with those of the mother and consequently this case supports the transmission of multiple maternal variants. Since it is the father with the most divergent variants it can be assumed that he was infected much earlier and probably infected his spouse with a minor variant, which has also undergone selective pressure over the years. In family 375, the appearance of child HIV-1 variants closely related to antenatal maternal sequences at 55 months of age possibly supports the hypothesis of the reappearance of the founder virus later on during infection.
Phylogenetic analysis of family 567 sequences
The mother's sequences were closer to those of the second sibling’ while the first sibling's sequences were further apart. Family sequences were supported by a bootstrap value of 86% (see Fig. 2d).
Discussion
In the absence of antiretroviral therapy, studies have shown that HIV-1 replicative fitness, which in turn impacts virus transmission, is largely determined by the functions of the envelope gene that was genotyped in our study. 40 –42 This is the first report attempting to assess HIV-1 subtype C envelope gp120 C2-V5 phylogenetic relatedness among close contacts in Zimbabwe following concurrent heterosexual and subsequent vertical transmission. In SSA where HIV prevalence is high, it is culturally acceptable for men to have more than one wife and/or extramarital sexual relationships. Ironically, the underrepresentation of these male sexual partners' involvement in PMTCT programs in this setting where HIV-1 transmission is predominantly heterosexual has compromised holistic HIV control strategies.
In 80% of heterosexual transmission cases, single viruses have been shown to establish infection 9,43 –45 with women harboring homogeneous env sequences being less likely to transmit per sexual act. 46 Limited heterogeneity observed in our mothers suggests that a single variant could have been acquired from the local site of infection. This could be a gender difference during pregnancy of HIV-1 subtype C possibly influenced by hormonal balances. In our study the directionality of heterosexual transmission, whether it was female to male (FTM) or male to female (MTF), could not be ascertained. Some subtype C studies have demonstrated similar FTM and MTF transmission rates. 47 Semen-derived viral populations have been shown to exhibit lower genetic diversity relative to the blood variants and this could probably be the reason for the limited heterogeneity observed in the mothers. 48 It could be worthwhile to explore similar but larger studies with many clones from the plasma and other compartments.
Contrary to our findings, some studies have demonstrated that women are often infected with multiple variants. 20 Diverse virus population in such studies could be attributed to reinfection by multiple partners since this other study's population was a cohort of female sex workers in Nairobi, Kenya with predominant A and D and to a lesser extent C HIV-1 subtypes. In the Kenyan study none of the five men and only two of 32 women had HIV-1 subtype C and interestingly both women had homogeneous virus populations. Homogeneous subtype C viruses have also been described elsewhere. 49,50 This is suggestive of the fact that subtype could influence the pattern of viral transmission.
Differences between the two studies could be due to variations in the study populations, the exact env region being analyzed, possibly variations in the sampling times at different stages of HIV-1 infection, and also different subtypes. Studies have shown that heterosexual and vertical transmission of HIV-1 subtype C viruses spread more rapidly due to increased mucosal and vaginal shedding. 51 –53 Compared to other subtypes, subtype C has also been shown to replicate and be transmitted more efficiently. 54 –56 Hence, there is a need to address subtype C-specific research questions rather than extrapolating and applying subtype B findings if curbing of the pandemic is to be realized.
Similar to previous observations a low degree of maternal HIV-1 genetic heterogeneity has been shown to correlate with vertical transmission contrary to observations by others. 31,57,58 If diversity remained low, then it could be likely that fewer variants could be harbored and hence there could be a narrow breadth of maternal neutralizing antibodies associated with increased risk of vertical transmission. Other important factors could be associated with infant exposure such as diverse viral inoculum, including other maternal factors such as nutritional status, human leukocyte antigen (HLA) genotype, coreceptor expression, or the presence of STIs among others. Successful transmission of maternal escape mutants has been reported in children sharing HLA alleles with their mothers. 59 Coinfection with human simplex virus type 2 (HSV-2) at delivery has been associated with increased intrapartum transmission of HIV-1. 60
Smaller genetic distances indicate a close genetic relationship whereas large genetic distances indicate a more distant genetic relationship. Our observation of distant first siblings' sequences is consistent with previous studies where viral divergence has been found to increase over time in children. 11 The first siblings were much older, about 60 months old, hence their HIV-1 could have evolved further from the maternal HIV, which was more or less closer to that of the second siblings who were about 15 months old.
The route of transmission may influence the genetic diversity, with some authors postulating that HIV homogeneity and heterogeneity patterns are likely to be different between infants and adults due to different exposures, different viral dynamics and set points following infection, and different immunity maturities in the recipients. 61 Studies have shown that most HIV-infected infants have a deficiency in cytotoxic T lymphocyte (CTL) responses to HIV 62,63 and inadequate CD4 T cell help. 64,65 Infection of infants with maternal CTL escape variants may further compromise the infant's ability to contain the virus. 66 On a positive note diversity may also disrupt the function of the env gene resulting in attenuation of HIV-1 virulence probably resulting in long-term nonprogressors in the absence of antiretroviral therapy observed in these pediatric patients. 67,68 Differences in mother–infant pair viral heterogeneity may also be due to virus compartmentalization between the plasma and breast milk variants. The immunological milieu of breast milk has been found to be distinct from that in blood as it contains a high concentration of HIV-1-specific T cells, antibodies, chemokines, and innate factors that modulate HIV-1 transmission risk. 69 –71 However, a subtype C study of breastfeeding mothers found no differences between breast milk and blood variants. 72
Families' subtype C intermingled with subtype C sequences from other regions, confirming the difficulty of classifying subtype C sequences on a geographic basis. 73 Due to the limited number of clones sequenced per family member it was not possible to determine the minor or major variants. More so, the directionality of heterosexual transmission could not be ascertained. It could be worthwhile to explore the determination of directionality of transmission using glycosylation patterns and amino acid lengths of HIV-1 env variable regions.
Few subtype C studies have looked at variation of the V1-V2 on transmission. 74,75 It would have been more interesting if the whole env region was sequenced including the V1-V2 region for comparison. However, sequencing of longer fragments has its own challenges. Phylogenetic analysis performed for each family sequence set suggested that several mechanisms may be involved in both vertical and heterosexual transmission. The star-shaped families tree suggested a localized expansion of the subtype C epidemic at least among these our families. Generally families' sequences clustered quite closely with sequences from South Africa, Malawi, and Botswana. Paternal sequences exhibited the most heterogeneous viral quasispecies while maternal sequences were relatively homogeneous. First siblings viral sequences were distributed into several branches suggesting multiple distinct lineages probably as a result of the evolution away from maternal viral sequences through immune selection pressures during their 5–6 years of life while second siblings' HIV-1 sampled between 12 and 15 months of age had relatively homogeneous viral populations closely related to maternal variants. Larger studies are warranted to address the caveats of this study and build upon its strengths. Our study could be the beginning of a family-based intervention in HIV research in Zimbabwe.
Sequence Data
Sequences were submitted to GenBank and the accession numbers assigned were JQ070719–JQ070752.
Footnotes
Acknowledgments
We gratefully acknowledge the families who participated in this study and support staff for facilitating the logistics. We also wish to thank collaborating institutions for capacity building and technology transfer: the University of Zimbabwe, Letten Foundation Research Centre, University of Oslo, and Oslo University Rikishospitalet. Special mention goes to the Letten Foundation and professor Letten F. Saugstad for funding the study.
Author Disclosure Statement
No competing financial interests exist.