
Abstract
Treatment of HIV-1 infection has been highly successful with small molecule drugs. However, resistance still develops. In addition, long-term use can lead to toxicity with unpredictable effects on health. Finally, current drugs do not lead to HIV-1 eradication. The presence of the virus leads to chronic inflammation, which can result in increased morbidity and mortality after prolonged periods of infection. Monoclonal antibodies (mAbs) have been highly successful during the past two decades for therapy of many diseases, primarily cancers and immune disorders. They are relatively safe, especially human mAbs that have evolved in humans at high concentrations to fight diseases and long-term use may not lead to toxicities. Several broadly neutralizing mAbs (bnmAbs) against HIV-1 can protect animals but are not effective when used for therapy of an established infection. We have hypothesized that HIV-1 has evolved strategies to effectively escape neutralization by full-size antibodies in natural infections but not by smaller antibody fragments. Therefore, a promising direction of research is to discover and exploit antibody fragments as potential candidate therapeutics against HIV-1. Here we review several bnmAbs and engineered antibody domains (eAds), their in vitro and in vivo antiviral efficacy, mechanisms used by HIV-1 to escape them, and strategies that could be effective to develop more powerful mAb-based HIV-1 therapeutics.
Introduction
S
One of the difficulties in developing mAbs as HIV-1 therapeutics is the extreme variability of the virus and the rapid emergence of resistant mutants. 2 This requires that antibodies exhibit a sufficient level of breadth in their ability to neutralize genetically diverse HIV-1 isolates. Several broadly neutralizing mAbs (bnmAbs) are highly effective against HIV-1 infection in vitro, 3 but their administration to HIV-1-infected humans has resulted in only modest antiviral effects. 4 In particular, viral rebound occurs in all patients receiving either high or low doses of bnmAbs, suggesting that virus variants resistant to the antibodies are rapidly generated and the antibodies may have difficulty in eliminating the virus and virus-infected cells in the densely packed lymphoid environment such as spleen, gut, and lymph nodes where HIV-1 mostly replicates and spreads. Therefore, novel potent antibodies are needed that exhibit broader neutralizing activity, are more resistant to HIV-1 escape, and may penetrate tissues better than the antibodies of conventionally used formats [e.g., immunoglobulin G (IgG), antigen-binding fragment (Fab), and single-chain variable fragment (scFv)]. Novel bnmAbs have recently been reported that may have potential as therapeutics and prophylactics in combination with other drugs. 5 –7
HIV-1 has evolved strategies to escape full-size antibodies and rapidly develop resistance. We, therefore, have hypothesized that antibody fragments of smaller size could be more effective than full-length antibodies. We have recently identified a small engineered antibody domain (eAd), designated m36, from a large phage-displayed human eAd library. 8 It targets a highly conserved sterically restricted structure on gp120 and potently neutralizes HIV-1 isolates from different clades. EAds are of very small size (11–15 kDa) while differing from small molecule inhibitors by retaining high specificity and, therefore, may have properties (e.g., access to sterically hidden epitopes and good penetration) to evade mechanisms used by HIV-1 to escape neutralization better than the antibodies generated by the human immune system. These new and other findings highlight the potential of mAbs as candidate HIV-1 therapeutics.
BnmAbs and Their In Vitro and In Vivo Efficacy Against HIV-1 Infection
HIV-1 entry is triggered by interaction of the viral envelope glycoprotein (Env) gp120 with cellular receptor CD4. 9 Binding of CD4 induces extensive conformational changes of gp120 that lead to coreceptor binding and release of Env gp41; the latter undergoes structural rearrangements to draw the viral and the cell membrane together, initiating fusion and enabling viral entry. These steps that are vital for infection are, therefore, the potential targets for bnmAbs (Fig. 1). 4,10
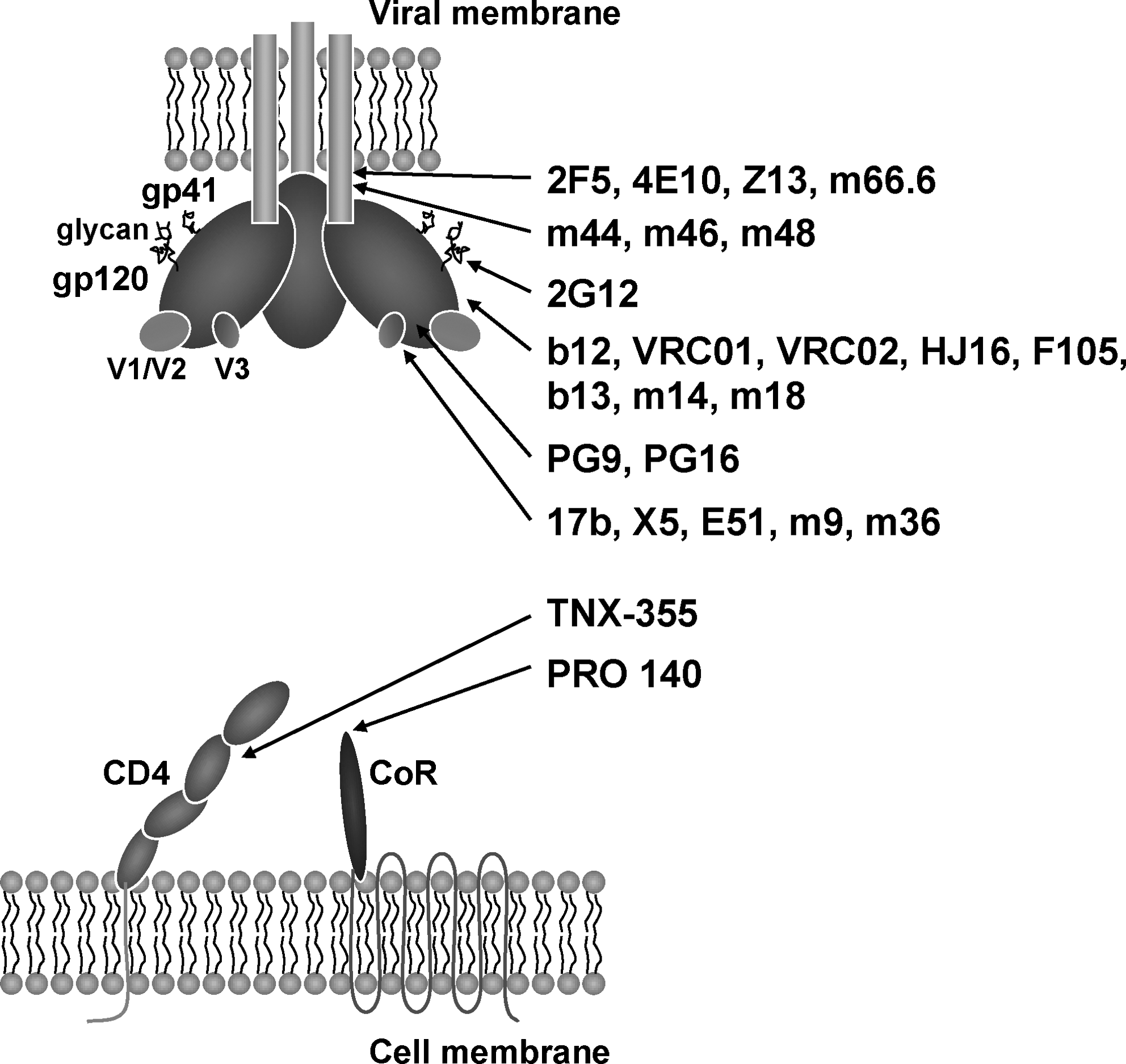
HIV-1 neutralizing monoclonal antibodies (mAbs) and their targets. HIV-1 entry is associated with binding of trimeric Env to the receptor CD4 and subsequently to the coreceptor on the target cell surface that results in virus–cell fusion. These steps and components of the fusion complex are potential targets for neutralizing antibodies. Several well-characterized neutralizing mAbs to each target are listed as representatives and discussed in this review.
BnmAbs to the CD4-binding site (CD4bs) on gp120
B12 is the first reported representative of the bnmAbs that target the CD4bs on gp120 as a competitive inhibitor of CD4 binding. 11 It was selected by phage display of an antibody library constructed from the bone marrow of an HIV-1-infected donor. Because CD4 binding is critical for infection and the CD4bs is functionally conserved, b12 is capable of efficiently neutralizing a wide range of HIV-1 isolates from different clades in vitro. 3 However, two other mAbs, F105 and b13, that also interact with the CD4bs are only weakly neutralizing. Recent structural analysis comparing b12 to F105 and b13 shows that although they approach gp120 at an angle that is very similar to that of CD4, slight differences in recognition result in substantial differences in F105- and b13-bound conformations relative to b12-bound gp120, suggesting that very precise targeting is needed for CD4bs antibodies to be broadly neutralizing. 12 Another two CD4bs mAbs, m14 13 and m18, 14 were selected from an immune human Fab library constructed from bone marrow obtained from three long-term nonprogressors whose sera exhibited the broadest and most potent HIV-1 neutralization among 37 HIV-1-infected individuals. They exhibited relatively broad and modest neutralizing activities against primary HIV-1 isolates representing different clades and inhibited cell–cell fusion.
The antiviral activity of b12 has been assessed in animal models. Mice with severe combined immunodeficiency (SCID) that were transplanted with human peripheral blood lymphocytes (hu-PBL) were injected with either IgG1 b12 or Fab b12 prior to challenge with HIV-1SF2. Fab b12, tested at a dose of 1.9 mg kg–1, was able to protect 25% of hu-PBL-SCID mice from HIV-1 infection while dose-dependent complete protection was achieved when the animals were administered IgG1 b12. 15 B12 was further tested in nonhuman primates. Intravenous infusion of b12 conferred dose-dependent protection to macaques vaginally challenged with an R5 simian/human immunodeficiency virus (SHIV). 16 B12 applied vaginally as a topical microbicide could also prevent SHIV infection in macaques. 17 These results provide evidence that treatment with bnmAbs as drugs or microbicides can prevent virus attachment and entry into the vagina and its associated tissues, thus representing a useful step in the development of an effective method to prevent sexual transmission of HIV-1 to humans. A recent study looked into the role of antibody Fc-mediated effector functions in the protection of macaques against vaginal challenge. 18 A significant decrease in the ability of b12 to protect macaques against SHIV challenge was observed when Fc receptor and complement-binding activity were engineered out of the antibody, whereas no loss of antibody protective activity was associated with the elimination of complement binding alone, suggesting the importance of antibody activity against both free viruses and virus-infected cells for effective protection. F105 was evaluated in a phase I human clinical trial. 19 Although it was well tolerated and nontoxic as a single intravenous injection, there was no obvious anti-HIV-1 activity observed.
In 2010, about 20 years after the discovery of b12, two novel CD4bs mAbs (VRC01 and VRC02) were reported that neutralized over 90% of circulating HIV-1 isolates with high potency in vitro. 7 VRC01 and VRC02 were identified by isolating single mAb-producing B cells with an antigenically resurfaced HIV-1 gp120 where the neutralizing surface of CD4bs was preserved while other antigenic regions were eliminated. Both antibodies closely mimic CD4 in binding as they also induce conformational changes in gp120 leading to enhanced exposure of the coreceptor-binding site. VRC01 has been shown to provide sterilizing protection of nonhuman primates from mucosal SHIV challenge, suggesting the potential usefulness of this antibody for vaccine development, therapy, and prophylaxis. 20 Another mAb, HJ16, recognizes an epitope overlapping the CD4bs. 6 It competes with sCD4 and b12 in binding to gp120s, and exhibits potent and selective neutralization of Tier-2 viruses.
BnmAbs to carbohydrates on gp120
BnmAb 2G12 was selected from hybridomas of human peripheral blood B lymphocytes from HIV-1-positive volunteers; it targets a conserved cluster of oligomannose glycans on gp120. 21 Binding of 2G12 could be abolished when the N-linked carbohydrates in the C2, C3, C4, and V4 were removed from gp120. 22 The “glycan shield” on gp120 is typically nonimmunogenic; antibodies that are able to block 2G12 binding were not detected in 16 HIV-1-positive human serum samples suggesting the 2G12 epitope is unusual. Because the mannose-dependant epitope of 2G12 is highly conserved, this antibody potently and broadly neutralizes primary and T cell line-adapted HIV-1 isolates (except clade C isolates) and inhibits syncytium formation in cell lines. Therefore, 2G12 provides proof of concept that carbohydrates on gp120 can serve as a target for bnmAbs.
2G12 alone or in combination with other bnmAbs has been tested in animal models and human clinical trials against either viral challenge or established infection. In an early experiment, 2G12, when applied alone intravenously, did not completely protect macaques from intravenous challenge of SHIV89.6PD, although the treated animals displayed a less profound drop in CD4+ T cells. 23 However, the antibody could be protective when the animals were vaginally challenged. 24 In line with this finding, a recent study showed that three of five rhesus macaques administered a low dose of 2G12 intravenously were protected with sterilizing immunity against vaginal challenge of SHIVSF162P3; one animal showed a delayed and lowered primary viremia and the other animal developed apparent signs of infection. 25 These results are in contrast to the previous finding that high titers of bnmAbs including b12 are typically needed for sterile protection in animals, suggesting the possibility that some epitopes on gp120 may be better targets than others. These results also suggest that antibody could better affect viral transmission through the vagina and associated tissues. Because in vitro studies 26,27 demonstrated that 2G12 could synergize with other bnmAbs including 2F5 and 4E10 in their neutralizing potency, they were evaluated in combination in animal models and HIV-1-infected persons. This will be discussed in the following section.
BnmAbs to the membrane-proximal external region (MPER) of gp41
Unlike the antibodies targeting gp120, two bnmAbs to gp41, 2F5 and 4E10, and a less potent one, Z13, bind to highly conserved linear epitopes at the base of gp41, the MPER. 28 In addition, 2F5 and 4E10 also interact with phospholipids on the viral membrane, which leads to stronger binding of the antibodies to the MPER and is required for neutralization. 29 Although the epitopes of 2F5 and 4E10 on the MPER do not overlap with each other, they both exhibit great breadth in their ability to neutralize genetically diverse HIV-1 isolates. 3 The epitope of Z13 lies between and overlaps with those of 2F5 and 4E10 but is closer to that of 4E10. 28 However, Z13 is not as broadly neutralizing as 4E10. The matured version of Z13, Z13e1, which has affinity comparable to that of 4E10, is still about an order of magnitude less potent than 4E10 against a variety of primary isolates. 30 In a pseudovirus-based comprehensive neutralization analysis of a small panel of HIV-1 mAbs, 4E10 exhibited broader interclade activity than others by neutralizing all the 90 isolates tested, although with relatively moderate potency; 2F5 neutralized 67% of the isolates but none from clade C; b12 neutralized 50% of the viruses from almost every clade; and 2G12 neutralized 41% but none from clades C and E. 3 Because only b12 and 4E10 were effective against viruses from clade C, these data provide implications for passive-immunization studies in countries in which clade C viruses are common. Recently, another MPER-targeted mAb, m66.6, was reported that binds to essentially the same epitope as 2F5. 31 M66.6 showed broad neutralizing activity, although to a lesser extent compared with 2F5. Interestingly, both the heavy and light chains of m66.6 originated from different germlines and are less mutated than 2F5, suggesting that neutralizing responses against the 2F5/m66.6 epitope can be elicited through various pathways.
Previous studies showed that when several bnmAbs targeting different epitopes were tested in combination, synergistic effects on neutralization against an SHIV were observed between any two of the antibodies. 26 Synergy of the combination regimens implies that combinations of antibodies may have a role in passive immunoprophylaxis against HIV-1. Therefore, the in vivo antiviral activity of 2F5 and 4E10 was mainly evaluated in combination with anti-gp120 mAbs in animal models and HIV-1-infected individuals. In an animal experiment, one of three macaques given a 2F5/2G12 combination intravenously 24 h prior to intravenous challenge by SHIV89.6PD exhibited only transient evidence of infection; the other two had a marked reduction in viral load. 23 A phase I clinical trial with the 2F5/2G12 combination was conducted on seven HIV-1-infected patients. 32 The antibodies were well tolerated. Transient reductions in viral loads and increases in CD4+ T cell counts and CD4+/CD8+ cell ratios were observed in five of seven and all individuals, respectively. However, virus escape from 2G12 occurred. More antibodies, including b12, F105, and 4E10, were then added to the regimen. Eleven of 16 newborn macaques were completely protected by triple or quadruple combinations of the antibodies b12, 2G12, 2F5, and 4E10 from oral challenge with different SHIV strains. 33 In a separate study, all four pregnant macaques treated with a triple combination of 2F5/2G12/F105 were protected against intravenous SHIVvpu+ challenge after delivery; the infants receiving the antibodies after birth and challenged orally with SHIVvpu+ shortly thereafter showed no sign of infection during 6 months of follow-up. 34 Furthermore, two of four macaque infants challenged orally with SHIV89.6PD and then given postexposure prophylaxis with a quadruple combination of b12/2G12/2F5/4E10 showed no evidence of infection; the other two maintained normal CD4+ T cell counts. 35 These results suggest the potential of bnmAb cocktails to prevent HIV-1 transmission through breastfeeding. The combination of 2F5/4E10/2G12 was chosen for human clinical trials. In a phase I evaluation, seven individuals showed no drug-related adverse effects. 36 During the phase II clinical trial, chronically and acutely infected individuals were recruited. 37 These subjects were on antiretroviral treatment (ART) before antibody administration. The capacity of the antibodies to suppress or delay the viral rebound after ART withdrawal was evaluated. The results showed that viral rebound was delayed in four of six acutely infected patients whereas only two of eight chronically infected ones showed a profound delay of rebound, suggesting that these bnmAbs could be more effective in early infection.
BnmAbs to the quaternary structures of trimeric gp120
Most of the bnmAbs described above exhibit relatively limited breadth and potency, especially with respect to nonclade B viruses, which account for the majority of infection outside North America and Europe; these antibodies are unable to completely eliminate viruses from infected humans; immunogens based on their epitopes have also failed to elicit broadly neutralizing immune responses. Therefore, novel approaches to isolate new bnmAbs that may be more potent, broadly neutralizing, and amendable to immunogen design are needed. Recently, two antibodies, designated PG9 and PG16, have been identified by functional high-throughput screening of about 30,000 activated memory B cells from a clade A-infected African donor. 5 PG9 and PG16 exhibited a high level of neutralizing activities and breadth comparable to or higher than those of the four bnmAbs, b12, 2G12, 2F5, and 4E10. Both antibodies preferentially reacted with trimeric gp120 than with monomeric gp120, indicating that their epitopes are highly dependent on the quaternary structure of the Env trimer. Mutagenesis analysis revealed that their epitopes are primarily located in the conserved regions of the V2 and V3 loops of gp120 and the preferential binding to trimeric gp120 is due to gp120 subunit presentation in the context of trimeric viral spike rather than gp120 cross-linking. These findings suggest that more conserved regions on the native Env trimer remain to be defined. Antibodies to these regions could better affect virus entry, have more potential as therapeutics, and are highly valuable for vaccine immunogen design. There are no reports to date about the in vivo activity of PG9 and PG16.
BnmAbs to the coreceptor-binding site (CoRbs) on gp120
The CoRbs on gp120 is highly immunogenic, eliciting many antibodies in vivo. It is exposed and/or formed after CD4 binding and, therefore, antibodies targeting this region are called CD4-induced (CD4i) antibodies. Compared to the bnmAbs described above, several CD4i mAbs including IgG1 17b 38 and X5 39 generally exhibit relatively less potent activity. An interesting finding with such antibodies is that their neutralization is inversely correlated with antibody size—Fabs (size, 50–60 kDa) neutralize viruses generally better than their IgG formats (size, ∼150 kDa for an IgG1) and scFvs (size, 25–30 kDa) could be even more potent than their Fab formats. 38 According to a previously published model, size-dependent neutralization could be due to steric restriction for antibody access to CD4i epitopes. 38 One of the reasons could be that after CD4 binds to the virus, the available space between the virus and the target cell surface is not sufficient to accommodate a whole antibody molecule but is adequate for antibody fragments. Thus, many CD4i mAbs that have been in development for use as potential therapeutics as single molecules or fusion proteins such as 17b, X5, and m9, 40 which is an improved version of X5, are in scFv or Fab format. Recently, m9 was tested against a large panel of isolates and showed superior activity compared to several well-known bnmAbs. 41 M36 is the first reported eAd (size, ∼15 kDa) that targets the CoRbs and potently neutralizes genetically diverse HIV-1 isolates in vitro. 8 In a humanized NOD/SCID/γc null mice model, m36.4, an affinity-matured version of m36, provided sterilizing protection in four of six animals against intrasplenical challenge with high-titer HIV-1 (>1000 TCID50s) while extensive infection was detected in all four control animals. 42
An obvious advantage for the use of antibody fragments is that they may penetrate better than full-length antibodies. This has been demonstrated with scFvs and Fabs, which move much faster in solid tumors than IgGs. 43,44 It has been hypothesized that antibody fragments may be better able to control HIV-1 infection because they are much smaller and therefore may be capable of avoiding certain defense mechanisms such as steric occlusion that HIV-1 has developed against antibodies generated by the host immune mechanisms and better able to penetrate tissues where HIV-1 replicates and spreads. 4 Unfortunately, there is a lack of data from in vivo experiments that could prove this hypothesis.
BnmAbs to the receptor CD4
In contrast to the antibodies to the CD4bs on gp120, a humanized mouse mAb, TNX-355 (Ibalizumab), was developed against the extracellular domains of human and rhesus CD4. 45 TNX-355 was engineered by grafting the complementarity-determining regions (CDRs) of its murine progenitor (known as 5A8) onto a human IgG4 construct to decrease possible immunogenicity and chances for CD4+ T cell depletion by antibody- and complement-mediated cytotoxicity. TNX-355 and 5A8 efficiently inhibited the in vitro infectivity of diverse primary isolates of HIV-1 probably by interfering with conformational changes on gp120, gp41, and/or CD4 that are required for virus– and cell–cell fusion, although they did not block binding of CD4 to gp120. 46 They also exhibited considerable antiviral activity in animal models. 47 Because TNX-355 and 5A8 do not target the major histocompatibility complex (MHC) binding site on the first extracellular domain of CD4, they do not interfere with the immunological functions of CD4. 48,49
A phase I human clinical trial with TNX-355 demonstrated that the antibody was well tolerated. A single dose of TNX-355 conferred a significant decrease in viral load and increase in CD4+ T cell counts in HIV-1-infected subjects.
50
A phase II study gave similar results, suggesting that the antibody was safe and effective in humans (
BnmAbs to the coreceptors
In addition to the receptor CD4, HIV-1 needs to use chemokine (C–C motif) receptor 5 (CCR5) (R5 virus) or another chemokine receptor CXCR4 (X4 virus) or both (R5X4 virus) as a coreceptor for entry, but the majority of primary HIV-1 strains use CCR5. This makes the coreceptors an attractive target for therapy. A humanized mouse anti-CCR5 mAb, PRO 140, 51 and its murine progenitor, PA14, 52 exhibited potent and broad-spectrum inhibition of primary HIV-1 R5 isolates from different clades while not affecting CCR5's chemokine receptor activity.
A phase I human clinical trial showed that PRO 140 given intravenously was generally well tolerated and exhibited potent, rapid, prolonged, and dose-dependent antiviral activity in the subjects with early-stage HIV-1 infection.
53
A phase II study has recently been completed that confirms the safety and favorable antiviral effects of PRO 140 (
Mechanisms that HIV-1 Could Have Evolved to Escape Neutralization by bnmAbs
HIV-1 has evolved multiple mechanisms in order to evade host immune responses such as rapid generation of variants and high variability and conformational masking of Envs. These have been studied 2 and discussed. 54 This review will focus on other possible mechanisms that could have immediate impact on the antiviral activity of therapeutic antibodies after infusion.
Mimicry of human self-antigens by conserved neutralizing epitopes on Envs that results in polyspecificity and autoreactivity of bnmAbs
Specificity is one of the major advantages of antibodies over small molecular inhibitors for targeted therapy. Some of the bnmAbs described above are from HIV-1-infected patients, very rare and valuable as candidate therapeutics and templates for the design of effective immunogens that could elicit the same or similar broadly neutralizing antibodies in vivo. However, previous studies demonstrated that 2F5, 4E10, and b12 were polyspecific against human self-antigens. In ELISA and Biacore-based assays, 2F5 and 4E10 bound to cardiolipin with relatively high affinity similar to those of autoimmune anticardiolipin antibodies. 29,55 2F5 also reacted with other antigens including histones and centromere B autoantigen, while 4E10 reacted additionally with the systemic lupus erythematosus (SLE) autoantigen. 55 Both 2F5 and 4E10 displayed lipid-binding properties. 56 B12 was positive against ribonucleoprotein, double-stranded DNA, centromere B, and histones. 55 The polyspecific autoreactivity was confirmed by interaction of the three antibodies with human HEp2 epithelial cells in a diffuse cytoplasmic and nuclear pattern as visualized by fluorescence microscopy, 55 and with human 293T, HOS, and SK-N-AS cell lines as demonstrated by flow cytometry. 57 2G12 was not autoreactive, but virus escape from 2G12 could easily occur. 58,59 The newly identified antibodies, PG9, PG16, VRC01, and VRC02, were tested against a panel of antigens and confirmed not to be polyreactive. 5,7
A previous study showed that antibody polyreactivity was associated with the CDR3 but not other regions of antibody heavy chains and could be fully preserved after antibody isotype switch. 60 2F5, 4E10, and b12 are unusual as they have long, hydrophobic heavy chain CDR3s. Mutation of hydrophobic residues in 2F5 and 4E10 heavy chain CDR3s abrogated lipid binding but had very little effects on binding of the antibodies to the MPER, also suggesting the role of heavy chain CDR3s in antibody polyspecificity. 61 Although these antibodies have been safely administered to a number of humans, it has been demonstrated that the autoreactivity of 2F5 enables induction of immunologic tolerance in knock-in mice, indicating the same possibility in humans. 62 Moreover, the antiviral activity of autoreactive antibodies could be significantly compromised by interactions with a variety of host self-antigens after infusion, which may partially explain why high doses of b12, 2F5, and 4E10 are typically needed for sterilizing protection in animals and confer only modest antiviral activity in humans. Thus, it is conceivable that HIV-1 could have evolved mechanisms to escape broadly neutralizing responses by having some conserved neutralizing epitopes as mimics of autoantibody epitopes.
Steric occlusion of conserved neutralizing epitopes on Envs that limits access of bnmAbs
Sterically restricted access of antibodies to the highly conserved CD4i epitopes, which overlap with the CoRbs on gp120, has been described above. In addition to the CoRbs, some conserved structures on gp41 are also presented in a sterically constrained environment. It has been demonstrated that smaller 4E10 fragments (e.g., scFv and Fab) are generally more potent in neutralization than its larger constructs (e.g., IgG and diabody), 63 which has not been observed with the CD4bs antibody, b12. In line with this observation, a polymeric IgM version of 4E10 was significantly less potent than IgG 4E10. 64 Moreover, 2F5 and 4E10 preferentially bind to a more “open” fusion-intermediate conformation of gp41 during its transient exposure 65 and neutralization by IgGs 2F5 and 4E10 could be potentiated by the addition of a peptide that holds the trimer in a prehairpin intermediate state after attachment of virus to cell surface. 66 These data provide supporting evidence for the sterically restricted access of antibodies to the MPER of gp41. During entry, gp41 undergoes a series of conformational changes that cause membrane fusion. Immediately prior to fusion, gp41 adopts a prehairpin intermediate in which the N-terminal (N-trimer) and C-terminal (C-trimer) trimers of gp41 are exposed. The human mAb, D5, binds to the conserved hydrophobic pocket on the N-trimer of gp41 and exhibits relatively broad neutralizing activity. 67 When converted to IgG1, D5 had up to a 12-fold reduction in neutralization potency over its corresponding scFv despite their slightly enhanced binding affinity. C37, which is a histidine-tagged version of the potent peptide inhibitor C34 targeting the N-trimer of gp41, showed a progressive loss of potency when fused to cargo proteins with increasing sizes from 6 to 41 kDa. 68,69 Use of a longer linker connecting C37 with cargo proteins could partially restore inhibitory potency. These results suggest that there is a steric hindrance with the N-trimer of gp41 in the prehairpin intermediate. All the physical constraints could significantly impair the intervention of some neutralizing antibodies passively administered or generated by the human immune mechanisms. Interestingly, gp41-specific mAbs targeting regions outside the MPER including m44, 70 m46, 71 and m48, 72 which exhibited relatively broad cross-reactivity but modest potency of neutralization, did not show significant dependence on size (M. Zhang and D.S. Dimitrov, unpublished observations). Some of these antibodies exhibited potency dependent on coreceptor concentrations—decreasing the cell-surface coreceptor concentration could increase the potency significantly (up to two orders of magnitude lower IC50s). 73
Encoding of immunogenic conserved nonneutralizing or enhancing epitopes on Envs leading to elicitation of nonneutralizing or enhancing antibodies that could antagonize bnmAbs
Neutralizing antibodies are the “good guys” in defense of infection. A number of HIV-1 neutralizing antibodies including several bnmAbs have been identified and characterized. However, the “bad guys” in the fight against HIV-1, enhancing and some nonneutralizing antibodies elicited by infection or Env-based immunization that could aggravate the progression of HIV-1 disease, are underexplored. Fc receptor-mediated (FcR-ADE) or complement-mediated (C-ADE) antibody-dependent enhancement of HIV-1 infection has been discussed previously. 74 Such types of enhancement were frequently observed in immunized or infected animals or humans. However, mAbs that could directly abrogate the beneficial effects of neutralizing antibodies were rarely discovered.
Previous studies showed that cross-reactive nonneutralizing antibodies (IgMs and IgGs) were elicited by immunizing mice with recombinant HIV-1 gp140s, suggesting the same possibility in humans. 75,76 Recently, we have successfully isolated several cross-reactive high-affinity nonneutralizing mAbs by panning a phage-displayed library constructed from an acutely infected patient (W. Chen et al., unpublished work). These antibodies target gp41, one of which significantly competes with a cross-reactive neutralizing antibody, m44, 70 in binding to gp140s. HIV-1-specific nonneutralizing antibodies were also selected from naive libraries constructed from healthy humans. 77 They are highly cross-reactive against gp120s and have high nanomolar affinity comparable to that of antibodies selected from immune libraries. Remarkably, some of the antibodies enhanced infection by HIV-1 primary isolates likely by stabilizing the CD4-induced conformations of gp120. Epitope mapping revealed that these antibodies target highly conserved structures on gp120, which are in very close proximity to the CD4bs and the CORbs. Because these two regions are vital for virus entry and harbor neutralizing epitopes, we have hypothesized that the selected nonneutralizing antibodies could directly block, by the mechanism of competition, the access of some neutralizing antibodies generated by the human immune system, especially those targeting the CD4bs or CORbs. This was supported by our finding that one of the antibodies strongly competed with the CD4bs antibody, b12, and partially reversed the neutralization by a CD4i antibody, m36. These results suggest that HIV-1 Envs have encoded antigenic conserved nonneutralizing or enhancing epitopes on both gp120 and gp41 that could be highly immunogenic in vivo to elicit “bad” antibodies to antagonize “good” antibodies, which may represent a strategy used by the virus to escape neutralizing responses.
Strategies to Increase the Potency of bnmAbs
The vulnerability of the current mAbs, especially in dealing with established HIV-1 infection, emphasizes that next-generation antibody-based therapeutics needs to be able to circumvent the viral defense mechanisms. We therefore discuss several possible solutions that could be effective, although only the evaluation in HIV-1-infected patients could definitely prove it.
Development of new approaches to isolate more potent and broader neutralizing antibodies
The recent identification of PG9 and PG16 by functional high-throughput screening sets a good example for the development of new approaches to isolate more potent bnmAbs. 5 Isolated Envs that are currently widely used for antibody selection may not preserve the native structures on the viral spike and, therefore, antibodies selected by isolated Envs may not be as potent as those directly selected for binding to viral particles or from functional screening. Previous efforts have been made to preserve or reconstruct the functional trimeric Envs, immunization with which resulted in improved elicitation of cross-reactive neutralizing antibodies against primary isolates, suggesting the usefulness of such structures for the selection of novel potent neutralizing antibodies. 78,79 The stabilized fusion intermediates formed during viral entry may also be useful because they could include highly conserved structures, as have been targeted by peptide inhibitors. 68,69 These structures could also be targeted by antibodies.
HIV-1 Envs are large proteins. They recognize several types of antibodies including isolate-specific nonneutralizing antibodies, some of which compete with cross-reactive neutralizing antibodies in binding to Envs. Therefore, the use of original Envs for selection could result in an unpredictable loss of some bnmAbs due to their relatively low affinity, modest expression levels, or competition with either isolate-specific or nonneutralizing antibodies with high affinity and expression. Based on this concept, a modified gp120, RSC3, was designed that specifically reacts with antibodies directed against the CD4bs. 7 The use of RSC3 led to the discovery of exceptionally potent and broad antibodies, VRC01 and VRC02. This approach can be extended to select novel bnmAbs against other highly conserved functionally important epitopes.
A lesson learned from the polyspecificity and autoreactivity of 2F5, 4E10, and b12 is that newly identified antibodies with potent and broad in vitro activity should be systemically evaluated for specificity in in vivo environments. It could be suggested that selected antibodies are tested with a wide range of human self-antigens, especially those that are dominant in human serum or other antibody-permeable tissues, as has been done to PG9, PG16, VRC01, and VRC02. 5,7 Specificity surveillance could also be conducted during panning where selected human self-antigens are added for depletion of unwanted antibodies.
Moreover, there could be a need to evaluate the new antibodies for possible competition by “bad” antibodies in neutralization. Many experimental and clinical data indicate that a proportion of the antibodies that develop early and persist throughout the course of HIV-1 infection includes “bad” antibodies of the immune system. 74 Instead of eliminating the virus, these antibodies facilitate its production in the body of infected persons by different means including antagonizing neutralizing antibodies. The reported 75 –77 and more “bad” antibodies that are to be identified could be used as reagents to further characterize the neutralizing antibodies of interest.
Development of bnmAb-based multifunctional entry inhibitors to combat viral resistance
It has been proposed that combining multiple entry inhibitors with different mechanisms of action would increase the antiviral potency and durability to resistance. In line with this proposal is the finding that a combination of gp120 and gp41 antibodies conferred more profound antiviral activity in vivo than either alone. 32 –35 Recently, a novel class of bifunctional fusion inhibitors (BFFIs) has been designed that consists of an anti-CD4 mAb (TNX-355 or 6314) covalently linked to a peptide fusion inhibitor (T-651 or its variants) at the C terminus of antibody Fc. 80,81 The BFFIs demonstrated more than 100-fold greater antiviral activity than the CD4 mAb or the peptide alone. They also displayed antiviral potency against the viruses resistant to other fusion inhibitors. In vivo pharmacokinetic studies showed that the BFFIs were stable in monkey and a dose of 10 mg kg–1 maintained serum concentrations greater than 2000-fold over the IC90 value for 7 days postdosing. 80 Another type of BFFI takes advantages of the synergy between CD4 and CD4i antibodies, which has been observed in several studies. 8,82 A fusion protein of the first two domains of CD4 (soluble CD4, sCD4) with 17b exhibited very broad and potent activity superior to that of b12, 2F5, 4E10, and 2G12 in neutralization against a large panel of isolates. 83 Joining of other CD4i mAbs (e.g., m36 and E51) to sCD4 also resulted in very potent and broad reagents. 84,85 Interestingly, a sCD4 fusion protein with a non-CD4i antibody also results in a potent reagent with more broadly neutralizing activity than either partner alone. 84 These results suggest that mAb-based BFFIs or multifunctional fusion inhibitors (MFFIs) could have improved potency and favorable pharmacokinetic properties and, therefore, offer a novel approach for HIV-1 therapy. However, whether they could be more durable to viral resistance remains to be proved.
Decreasing the molecular size of bnmAbs to circumvent the physical constrains of Envs
Two fundamental problems with large full-size antibodies and their fusion proteins are their poor penetration into tissues (e.g., solid tumors) and poor or absent binding to regions on the surface of some molecules (e.g., HIV-1 Envs) that are accessible by molecules of smaller size. It has been proposed that further decreasing the size of antibodies to eAds could lead to exceptionally potent neutralizers. In addition to better penetration, eAds could be better able to control virus replication because they could approach more conserved hidden epitopes. Such epitopes could be more invariable as required for the maintenance of biological functions (e.g., substantial binding to the receptor or coreceptor) and, therefore, the antibodies could be more resistant to viral escape. We have recently demonstrated that a human eAd (size, ∼15 kDa), m36, targets a highly protected structure (CD4i epitope) on gp120 and exhibits exceptionally potent neutralizing activity against diverse HIV-1 primary isolates, with potency on average comparable to or higher than those of the bnmAb, scFv m9, and the inhibitory peptide, C34. 8 Increasing antibody size by joining to cargo proteins could lead to complete blocking of neutralization, suggesting the existence of steric occlusion with the highly conserved functionally important structure that the antibody targets. Several single heavy chain variable domains (referred to as VHHs) selected from llama immunized with HIV-1 gp120 have been characterized as potent HIV-1 entry inhibitors, interfering with virus binding to CD4. 86
Two major issues need to be addressed before eAds can be suited for in vivo use. These include their short half-life in circulation and lack of biological effector functions as has been described for other antibody fragments, including scFvs and Fabs. A possible solution is to fuse eAds with human antibody Fc. Although this strategy brings eAds back to agents with medium molecular weight (∼75 kDa), they could still promise more efficient penetration than full-length antibodies (size, ∼150 kDa for an IgG1) and long half-life in vivo. Importantly, Fc-fusion proteins of eAds could preserve the better ability of targeting certain hidden conserved epitopes such as the CD4bs epitopes than full size antibodies; such epitopes could be better accessed by eAds that in general have smaller paratopes than the Fabs of full-size antibodies. This solution may not be applicable to eAds against CD4i epitopes. CD4i epitopes are sterically restricted structures formed during HIV-1 entry and, therefore, the increase in antibody size could dramatically diminish neutralization potency. 8 However, the accessibility problem can be solved by further fusing the CD4i eAd-Fc with sCD4, which helps expose the CD4i epitopes without involvement of the cellular CD4. 85 Recently, engineered soluble stable single human CD4 domains (D1s) have been successfully generated by combining the power of structure-based design with panning and screening large D1 mutant libraries against different HIV-1 Envs. 87 They (size, ∼12 kDa) are smaller than two-domain sCD4 (size, ∼25 kDa) and, therefore, can be used to decrease the molecular size of CD4i eAd-sCD4-Fc constructs. Other novel strategies that could retain the small size of eAds while offering effector functions and favorable pharmacokinetic properties could be more beneficial.
Conclusions
There are currently 29 mAbs approved in the United States and the European Union against various diseases including the only one, Synagis (palivizumab), for prevention of an infectious disease. No mAb-based therapeutics have been approved for therapy of infectious diseases. All bnmAbs tested in HIV-1-infected humans have failed to provide favorable clinical benefits, most likely because they are not capable of circumventing the escape mechanisms used by the virus. This is in contrast to the clinical benefits provided by the currently approved therapeutic antibodies (e.g., those targeting cancers); these obstacles appear to be a major reason why there are no such antibodies against HIV-1 even in phase III clinical trials. The disappointing results urge rethinking concerning the therapeutic potential of neutralizing antibodies in the treatment of HIV-1 infection. In addition to the challenges faced at the biochemical and molecular levels, the use of antibody-based therapies in routine clinical practice is also limited by several factors including relatively high costs and the disadvantages associated with drug administration. Many small molecule inhibitors that have been approved for treatment of HIV-1 are highly effective, can be orally administered, and are relatively cheaper than antibodies, although they show toxicities and encounter resistance after long-term use. From a positive point of view, however, antibodies are generally safe and provide passive protection, which still raises the general interest in developing antibody-based drugs against HIV-1 such as microbicides, which can be easily applied topically. With continually decreased manufacturing costs and increased serum half-lives that enable low-frequency administration, antibodies are particularly attractive as candidate HIV-1 prophylactics and therapeutics. If systematic and careful strategies are developed based on the increasing understanding of the interplay between the virus and the human immune system, the initial hope that HIV-1 could be eradicated in patients by mAb-based drugs may still be fulfilled.
Footnotes
Acknowledgments
We thank John Owens and Emily Streaker from our group for technical assistance and helpful comments. This project was supported by the Intramural AIDS Targeted Antiviral Program of the National Institutes of Health (NIH), by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research, and by the Gates Foundation (D.S.D.).
Author Disclosure Statement
No competing financial interests exist.