
Abstract
Human immunodeficiency virus (HIV) long terminal repeat (LTR) promoter-mediated gene expression is regulated by the viral Tat protein that relieves a block to viral transcription elongation after binding with a viral hairpin loop RNA structure called the trans-activation-responsive region (TAR). Tat protein significantly up-regulates viral genome transcription and hence it has long been considered a potential target for antiretrovirals. Here we report the construction of a plasmid containing an HIV-1 LTR-driven reporter cassette with a colinear tat gene under control of a viral promoter and thus conditionally configured for constitutive expression of reporter genes. Inhibition of luciferase reporter expression in a cell line harboring the plasmid in the presence of tat-targeted shRNA confirmed the specificity of the assay and a dose-dependent reporter activity inhibition by the fluoroquinoline derivative K-37, a class of small RNA binding molecule that inhibits Tat and other RNA-dependent transactivations, further validated the method. Subsequently we also made a lentiviral vector (LV) containing the same transcription units and derived a stable cell line using the said LV and similar dose-dependent inhibition was documented using K-37. This quick and sensitive reporter-based method is the simplest screening assay for putative inhibitors of HIV-1 Tat-induced LTR-driven gene expression requiring test material addition as the only manipulation.
T
Though it is well documented that Tat actually initiates assembly of a transcription elongation complex comprised of P-TEFb (a heterodimer composed of CDK9 and cyclin T1) and several other well-characterized host cellular cofactors, the exact mechanism of how each factor, and possibly still to be identified factor(s), acts stepwise to form finally the super elongation complex (SEC) that results in PolII-mediated viral transcription elongation is not yet completely understood. 8 –10 Inhibition of Tat function through interference with Tat-mediated viral gene expression thus provides an attractive alternative target for AIDS therapy. 11 –14 Putative antagonists are selected such that they interfere with the appropriate Tat complex–TAR interaction process with high avidity and selectivity, thereby competitively preventing the Tat-mediated trans-activation response.
Clear molecular understanding of this interaction led to the development of sensitive assays to find potential inhibitors without the use of infectious virus, and most methods used some known reporter protein to arrive at a semiquantitative measure of inhibitor efficiency. Assays based on similar transactivator and reporter constructs described earlier involved transient cotransfections, which required internal control reporter vectors to monitor inherent variations in transfection efficiency between experiments and/or to normalize transcriptional activity. 15,16 To reduce the number of experimental manipulations and assay time, we developed a very simple, one-step method. This included making an indicator cell line harboring a plasmid that houses an HIV-1 LTR-driven reporter construct with a colinear tat gene under control of the cytomegalovirus (CMV) immediate early promoter. The cell line constitutively expresses Tat protein thereby simultaneously activating the reporters through the LTR and thus becomes suitable for screening of potential antagonists, the addition of which is the only required experimental step.
Luciferase is a popular choice as a reporter because a functional enzyme is created immediately upon translation, and the assay is rapid, reliable, cheap, can be adapted to high-throughput applications, and is easy to perform. Enhanced green fluorescent protein (EGFP)-derived fluorescence is also a very user friendly reporter for imaging or fluorimetry/ELISA-based assays. However, a luciferase-based assay is a more sensitive and discernible dose-response indicator than EGFP, possibly due to the long half life (∼26 h) of EGFP. 17
To obtain the functional transactivator-reporter plasmid, the LTR.Luc-IRES.EGFP-PA fragment bearing the HIV-1 subtype C LTR was released by digestion with NsiI from the plasmid pLTR.Luc-IRES.EGFP (from D. Mitra) and subcloned at the BglII (polished) site of the plasmid pcDNA-Tat (from U. Ranga). 18,19 The resultant plasmid, pLG-tat, consists of the HIV-1 LTR promoter-controlled transcription of firefly luciferase gene-Luc and the EGFP gene followed by the CMV promoter-driven tat gene expression unit (Fig. 1a).
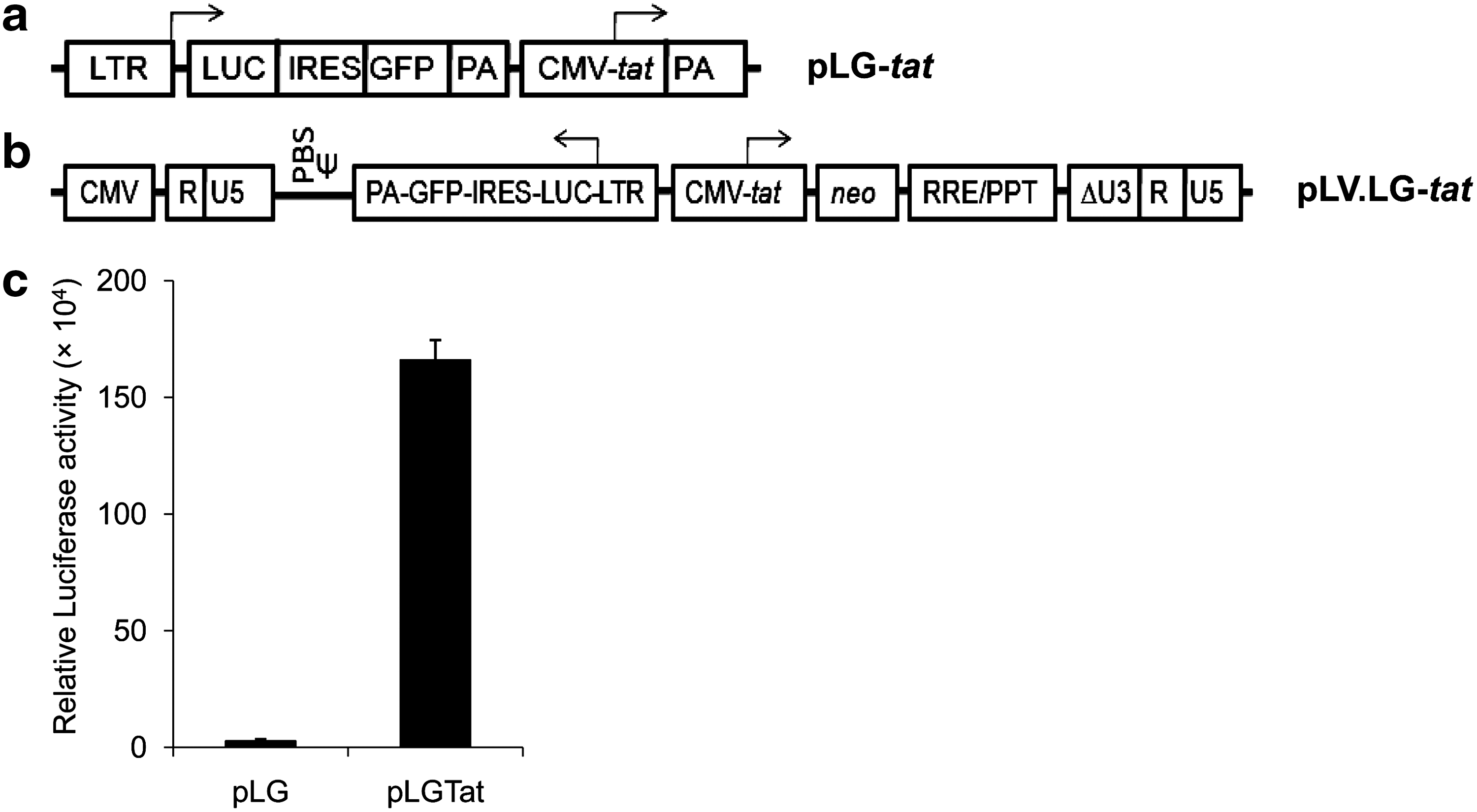
Genomic organization of the transactivator-reporter gene cassettes and Tat-induced reporter expressions. Configuration of the genes in
To establish a stable reporter cell line, the human embryonic kidney-derived cell line HEK-293, grown in media consisting of Dulbecco's modified Eagle's medium (Invitrogen Corporation, USA) supplemented with 10% fetal bovine serum (Invitrogen) and 50 μg/ml gentamicin (Nicholas-Piramal, India) at 37°C/5% CO2 atmosphere, was plated at 4×105 cells/60-mm culture dish (Nunc, Denmark) a day prior to transfection. Cells were transfected with pLG-tat using Lipofectamine 2000 (Invitrogen) following the manufacturer's instructions and sorted after 48 h in a cell sorter (FACS ARIA; BD Biosciences, USA) based on EGFP fluorescence and further selected in the presence of media containing 800 μg/ml G418 (Sigma, USA) and the stable cell line was GFP positive (Supplementary Fig. S1a) (Supplementary Data are available online at
Cells from the stable lines were cultured in a 96-well flat-bottom plate (Nunc) at a density of 5×103 cells per well in 100 μl medium for 16 h to determine reporter activity after 48 h using the Steady-Glo Luciferase assay system following the manufacturer's instructions (Promega, USA) and the luminescence signal was detected using a microplate reader (Mithras LB-940; Berthold, Germany). A significant increase of the luciferase activity (>80-fold) in the indicator cell line compared to the control one established the functional appropriateness of the indicator cell line (Fig. 1c).
We assessed the specificity of the assay system by shRNA-mediated tat down-regulation. A U6 promoter-driven shRNA cassette was generated by polymerase chain reaction (PCR), targeting nucleotides from positions 151 to 171 of an HIV-1 subtype C Tat-coding sequence (GenBank accession number FJ765005), yielding a U6-shRNA-polyT cassette; target sequence selection was based on published guidelines. 20 A control nonrelevant red fluorescence protein (DsRed) shRNA was also generated identically and both PCR products were cloned in a T/A cloning vector pTZ57R (MBI Fermentas, Lithuania). First the level of Tat protein expression was checked in the presence of the shRNA by transfecting 0.2×106 indicator cells, seeded the previous day in antibiotic-free media, separately with empty vector, vector containing DsRed shRNA, or the tat-specific shRNA using lipofectamine.
Cells were harvested after 48 h, lysed using Proteojet (MBI Fermentas), proteins were separated on a 15% SDS–PAGE, and the resolved proteins were immunoblotted using antiserum to HIV-1 tat (Cat# 705, AIDS Research and Reference Reagent Program). 21 Subsequently, tat-specific shRNA-mediated down-regulation of Tat-mediated transactivation was also tested on the indicator cell line. Indicator cells (2×103) in 100 μl antibiotic-free media were seeded in 96-well flat-bottom plates and transfected next day with 320 ng of DNA of empty vector or shRNA to DsRed or vector encoding shRNA to tat. Tat activity was measured as relative transactivation by luciferase assay 48 h posttransfection. A significant reduction of Tat protein expression as shown by immunoblotting as well as reduction of luciferase activity by the tat-shRNA, but not by DsRed-shRNA, clearly proved that down-regulation of reporter expression is subject to the specific disruption of Tat-mediated transactivation in the test cell line (Fig. 2a and 2b).

Effect of tat targeted shRNA.
After characterization of the plasmid-containing cell line, we also derived a cell line housing the same gene cassettes but delivered through a lentiviral vector (LV) by transduction. To obtain the said genes in effective configurations within a lentiviral transfer vector, first the CMV promoter-tat coding sequence was released from its parental plasmid by SalI/XbaI digestions and cloned at XhoI/XbaI sites of the pLV-neo, a neomycin selection marker bearing derivative of the third generation HIV-2-based LV reported earlier, to make pLV-tat-neo. 22 The LTR.Luc-IRES.EGFP-PA fragment was released by NsiI digestion from its parental plasmid and cloned at an identical site to pTZ57R. This fragment was further released by XbaI (polished)/SalI digestions and cloned in pLV-tat-neo at EcoRV/SalI sites to obtain the lentiviral transfer vector pLV.LG-tat (Fig. 1b). This plasmid, along with other packaging constructs, was used to generate virus particles in HEK293 FT cells following procedures as described elsewhere. 22 HEK 293 cells (4×105) were plated on a 60-mm culture dish (Nunc) and were transduced next day using the virus particles generated and cultured for 72 h followed by GFP fluorescence-dependent sorting and selection under G418 to obtain a stable GFP-positive cell line henceforth requiring no antibiotic selection (Supplementary Fig. S1b).
The efficacy and utility of the assay were evaluated using K-37 [7-(3,4-dehydro-4-phenyl-1-piperidinyl)-1,4-dihydro-6-fluoro-1-methyl-8-trifluoromethyl-4-oxoquinoline-3-carboxylic acid], a fluoroquinoline derivative that has a distinct inhibitory effect on RNA-dependent transactivation at nanomolar concentrations without modulating the host cellular factors. 23,24 Results from the structurally related compound suggest that this drug inhibits the TAT–TAR interaction most possibly interacting with the bulge region of the TAR. 13 The compound was dissolved in DMSO to make a 2 mM master stock. Then 10 μM working stock was prepared in culture media from the above and used at a final concentration of 0.25, 0.5, and 1.0 μM, respectively, for the luciferase assay; percent inhibition of luciferase activity was calculated in comparison to test result without the drug.
Simultaneously, an identical assay was carried out using azidothymidine, a reverse transcriptase inhibitor, as a negative control (AZT; Sigma). At increasing concentrations of K-37, a characteristic dose-dependent inhibition profile was obtained reaching 75% inhibition of luciferase activity at 1.0 μM, in both the cell lines, whereas AZT did not show any appreciable inhibition (Fig. 3a and b). Since drugs often inhibit the reporter activity at their cytotoxic concentrations, we evaluated the same for K-37 and AZT by MTT assay. Then 1×104 indicator cells were cultured in the presence of the indicated concentrations (μM) of K-37 and AZT in a 96-well plate for 48 h. Subsequently, 20 μl MTT (5 mg/ml; USB Corporation, USA) was added to each well, incubated for 4 h at 37°C, followed by the addition of 50 μl DMSO per well and 10 min incubation on a shaker. Absorbance was measured at 550 nm with 650 nm reference wavelength (Fig. 3c). K-37 did not show any overt cytotoxicity but significantly inhibited Tat-mediated gene expression implying that the cell viability variation was not necessarily concomitant with the observed reporter expression down-regulation.

Reporter bioassay profiles of the indicator cell lines in the presence of the drug K-37 and the effect of K-37 on cell viability and Tat protein. Inhibition of Tat-mediated luciferase reporter transactivation under different doses (μM) of K-37 and AZT in
Furthermore, the Tat protein expression profile was also evaluated in the presence of the same doses of K-37. Then 1×105 indicator cells were cultured in a 6-well plate for 48 h under the indicated doses of K-37 followed by preparation of cell extracts and immunoblotting as described for the shRNA experiment. No noticeable alteration of Tat level ruled out any possibility that the drug affects transactivator expression (Fig. 3d). Thus, the suitability of the assay system as a Tat-mediated transactivation inhibitor screening method was validated conclusively; a graphic abstract depicts the simplicity of the assay (Supplementary Fig. S2).
Existing methodologies for profiling drug-mediated inhibition of Tat–TAR interaction/ transactivation inhibition rely on the profiling of EGFP or enzymatic activity, commonly using firefly luciferase, chloramphenicol acetyltransferase (CAT), β-galactosidase, or secreted alkaline phosphatase (SEAP). 15,16,25 –30 Alternatively, an end point cytotoxicity-based assay was also developed that takes a week to accomplish. 31 The method described here ensures that the only single manipulation required is addition of the putative interfering drug and thus completely bypasses time-consuming transfections/cotransfections and the scope of any variations from there or the time required for a cell viability-based assay.
Inclusion of the SteadyGlo substrate in the assay allows long-term signal stability following initiation of enzymatic action and is thus convenient for multiple plate handling in high-throughput format and the GFP positivity of the indicator cells is a convenient monitoring guide to indicate the appropriateness of the cell lines for the assay. Use of two different classes of reagents, a specific shRNA and a proprietary drug, K-37, both showing similar end point profiles, confirmed the specificity of this assay. Moreover, the pLG-tat plasmid can be used to make an indicator cell line of any lineage as per the user's choice.
Alternate availability of a cell line with LV integrated indicator constructs offers a selection-free cell line. This infectious virion-free, rapid, cost-effective assay using a very small amount of reagents and cells is robust, sensitive, and thus adaptable to a high-throughput screening format to find novel compounds, targeted to inhibit Tat-mediated activation of HIV-1 replication, as an adjunct AIDS therapy modality.
Footnotes
Acknowledgments
The study was supported by a grant from “ICMR-DBT collaborative effort on HIV/AIDS and microbicides” to R.M. We thank Dr. Uday Ranga, JNCASR, Bangalore for the pcDNA-Tat plasmid and Dr. Debashis Mitra, National Cell Science Centre, Pune, for the pLTR.Luc-IRES.EGFP plasmid and valuable inputs to improve the manuscript. We thank Prof. Vinayaka R. Prasad, Albert Einstein College of Medicine, New York, for helpful suggestions.
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.