
Abstract
The Gag proteins of HIV-1 are central players in virus particle assembly, release, and maturation, and also function in the establishment of a productive infection. Despite their importance throughout the replication cycle, there are currently no approved antiretroviral therapies that target the Gag precursor protein or any of the mature Gag proteins. Recent progress in understanding the structural and cell biology of HIV-1 Gag function has revealed a number of potential Gag-related targets for possible therapeutic intervention. In this review, we summarize our current understanding of HIV-1 Gag and suggest some approaches for the development of novel antiretroviral agents that target Gag.
Introduction
The HIV-1 replication cycle
HIV-1,

Representation of an HIV-1 virion. The locations of the Gag proteins matrix (MA), capsid (CA), and nucleocapsid (NC), the viral enzymes reverse transcriptase (RT), protease (PR), and integrase (IN), and the Env glycoproteins are indicated. The figure was generously provided by L. Henderson, National Cancer Institute, Frederick, MD.
The HIV-1 infection process begins with the attachment of gp120 to the target cell plasma membrane (Fig. 2). 1 –4 The principal attachment receptor for HIV-1 and other primate lentiviruses is CD4. Productive infection also requires the presence of a coreceptor, typically CXCR4 or CCR5. The binding of gp120 to CD4 and coreceptor triggers conformational changes in gp41, which in turn lead to the fusion of the viral envelope and the target cell membrane and entry of the viral core into the host cell cytoplasm. Recent evidence suggests that HIV-1 entry can also occur in a low-pH endosomal compartment after receptor-mediated endocytosis. 5 Upon entry of the virion into the cytosol, the Env glycoproteins and the lipid-associated MA protein dissociate from the incoming particle at the membrane, and the poorly understood process of uncoating is initiated.
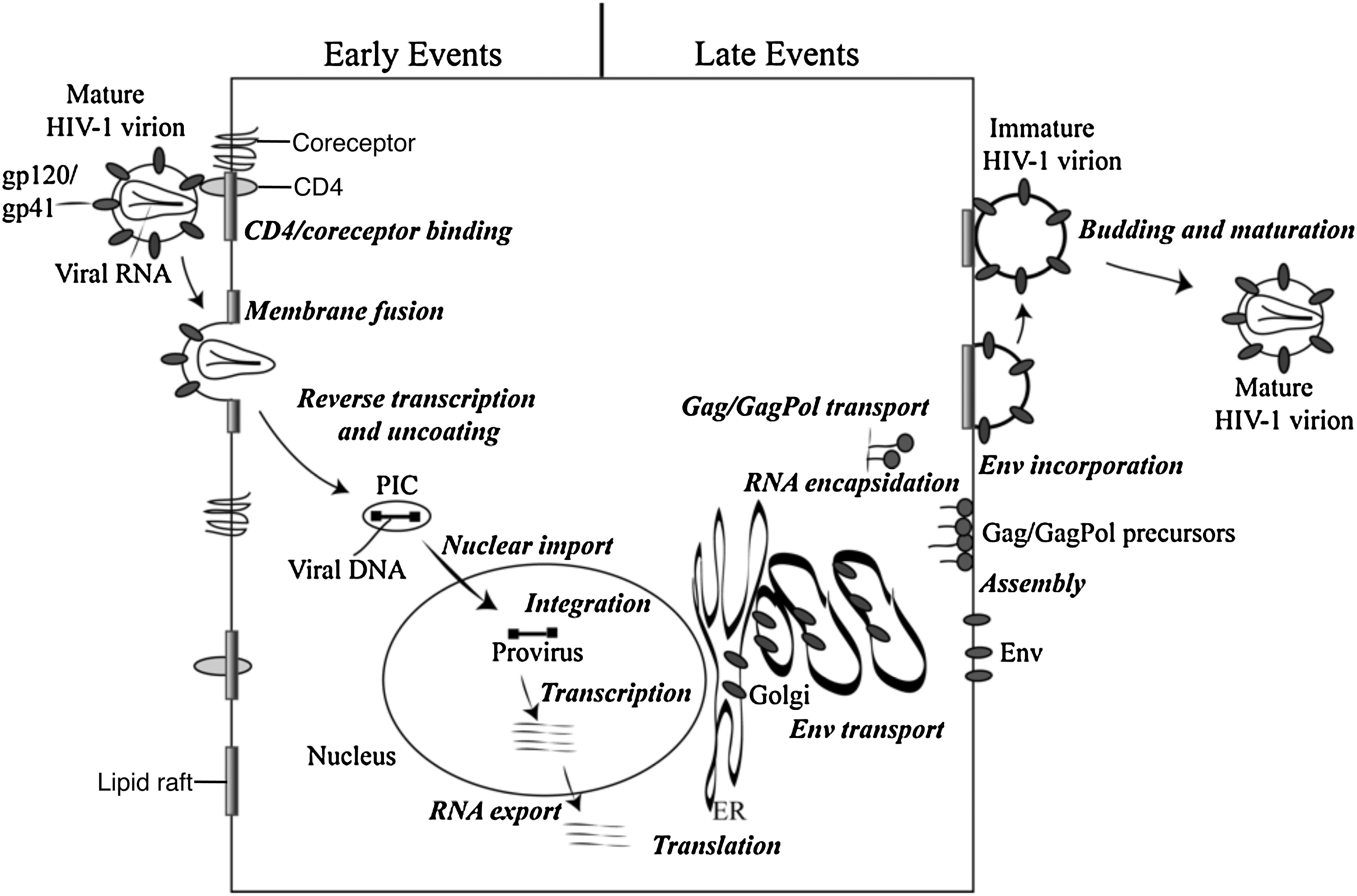
Schematic representation of the HIV-1 replication cycle. The details of the replication cycle are described in the text. Reprinted with permission from Freed (2004). 19 Copyright 2004 Elsevier Inc.
The enzymes RT and IN, together with the NC protein, remain in close association with the viral RNA as it is converted to double-stranded DNA by RT-catalyzed reverse transcription. 6 NC acts as a nucleic acid chaperone at several steps during reverse transcription to facilitate the conversion of RNA to DNA. 7 Vpr is also a component of the reverse transcription complex (RTC). The extent to which CA remains associated with the incoming RTC has been a topic of debate. However, reverse transcription and uncoating appear to be temporally linked, 8 and it is clear that some host restriction factors that block early postentry steps in the viral replication cycle target CA. 9,10 The newly reverse transcribed viral DNA is translocated to the nucleus in a structure known as the preintegration complex (PIC). The nuclear import process remains incompletely understood; however, a role for CA in this process 11,12 implies that some CA protein may remain associated with the viral nucleoprotein complex as it traffics to the nuclear pore. Once inside the nucleus, the double-stranded viral DNA integrates into the target cell genome through the action of the IN enzyme. 13 The integrated viral DNA serves as the template for transcription from the viral promoter in the 5′ long terminal repeat (LTR) to generate the spliced viral mRNAs and full-length genomic RNAs; these are transported out of the nucleus via the action of the Rev protein. 3
The Gag proteins are translated from full-length message as a polyprotein precursor containing MA, CA, NC, and p6 domains as well as two spacer peptides, SP1 and SP2. 14,15 During translation of the Gag precursor, known as Pr55Gag, an occasional 1 ribosomal frameshift leads to the production of a GagPol precursor protein (Pr160GagPol), the abundance of which is approximately 5% that of Pr55Gag. The Gag and GagPol precursor polyproteins are transported to the plasma membrane, where they assemble and incorporate the viral Env glycoproteins. The membrane targeting of Gag and GagPol is regulated by the MA domain, which also plays an important role in the incorporation of the viral Env glycoproteins. Assembly takes place in cholesterol-rich membrane microdomains (lipid rafts) through direct interactions between MA and the phospholipid phosphatidylinositol-4,5-bisphosphate [PI(4,5)P2]. 16,17 Interactions within the CA domain of Gag initiate the Gag assembly process.
The two copies of viral RNA are encapsidated during the assembly process via direct interactions between the genomic RNA and the NC domain of Gag. 7 NC–RNA interactions also play an important role in promoting Gag–Gag interactions. The nascent virus particle buds and pinches off from the plasma membrane by recruiting components of the cellular endosomal sorting machinery, specifically, the endosomal sorting complexes required for transport (ESCRTs) and associated factors. 18 –20 Upon release from the infected cell, the viral PR cleaves the Gag and GagPol polyprotein precursors in a highly ordered, step-wise processing cascade. Gag and GagPol cleavage triggers virion maturation. 21 In the mature virion, MA is associated with the inner leaflet of the lipid bilayer and CA reassembles to form a conical shell around the viral RNA in complex with RT, IN, and NC. With maturation, the viral replication cycle is complete and a new round of infection can take place.
Current status of antiretroviral therapy
Over the past three decades, HIV-1 infection has led to the deaths of more than 35 million people worldwide, with over 33 million currently infected (
There are currently ∼25 FDA-approved antiretroviral drugs available to treat HIV-1-infected patients. 22 The majority of these target two viral enzymes—RT and PR. RT inhibitors fall into two mechanistic classes, the nucleoside-analog RT inhibitors (NRTIs) and the nonnucleoside-analog RT inhibitors (NNRTIs). An inhibitor is also available that binds gp41 to inhibit fusion (e.g., enfuvirtide), and maraviroc, the first antiretroviral drug that targets a cellular protein, acts by binding CCR5 to prevent viral fusion and entry. In 2007, the first IN inhibitor, raltegravir, was approved. Because treatment with any single drug rapidly leads to the emergence of drug-resistant viral variants, patients are treated simultaneously with a cocktail of inhibitors, often referred to as highly active antiretroviral therapy (HAART).
Although HAART has been very effective in patients with access to therapy, drug resistance continues to be a serious problem that in many cases limits the effectiveness of available antiretroviral agents. 23 Drug accessibility, toxicity, and compliance issues have also blunted the positive impact of HAART. For these reasons, it is our opinion that the HIV-1 research community should remain actively enGaged in identifying additional drugs, particularly those that target novel steps in the replication cycle. Although PR inhibitors block Gag and GagPol processing by interfering with the enzymatic activity of PR, it is noteworthy that there are currently no drugs in clinical use that act on any aspect of Gag function. This review addresses the possibility that events in the HIV-1 replication cycle promoted by Gag—virus assembly, Env incorporation, genomic RNA encapsidation, particle budding and maturation, uncoating, and possibly nuclear import—constitute viable antiretroviral targets. Much of what we propose is highly speculative, but we hope it will help guide future efforts in this aspect of HIV-1 drug discovery and development.
Gag as a Target for Antiviral Therapy
The HIV-1 particle production pathway follows a series of discrete steps that include (1) targeting of Gag to the site of virus assembly, (2) binding of Gag to cellular membranes, (3) interactions between Gag monomers leading to multimerization, and (4) budding and release of nascent virions from the infected cell. As discussed above, HIV-1 particle production is driven by the Gag polyprotein precursor, Pr55Gag (which is often referred to as “Gag”), and each domain of Gag is involved in the assembly and release process. The MA domain directs Pr55Gag to the plasma membrane of the host cell where it anchors Gag to the inner leaflet of the lipid bilayer. The CA domain, together with SP1 and NC, homooligomerizes to drive the multimerization of Gag and promote particle assembly. The NC domain also binds to the viral genomic RNA, leading to the packaging of two copies of the viral genomic RNA into each virus particle. The C-terminal p6 contains motifs known as “late domains” that interact with cellular ESCRT machinery to promote the fission and release of virions from the cell surface. As mentioned above, cleavage of Pr55Gag by the PR leads to virion maturation. During the assembly process a number of cellular factors are incorporated into virions. It has been reported that ∼250 cellular proteins are present in HIV-1 virions produced from monocyte-derived macrophages. 24 In most cases, the functional significance of these virion-associated host proteins remains to be established.
The immature virus particle is composed of ∼5000 Gag molecules that are tightly packed to form a roughly spherical protein shell. 25,26 High-resolution cryoelectron microscopy (EM) analyses have demonstrated that rod-shaped Gag molecules are packed within immature particles side-by-side in a radial fashion, with the N-terminal MA domain associated with the membrane and the C-terminal portion of Gag oriented toward the center of the particle. 27 –29 In immature virus particles, Gag molecules form a continuous but incomplete hexameric lattice. 30,31
MA
The MA domain plays an important role in the trafficking of Gag to the assembly site, which in most cases is the plasma membrane. Structural analyses revealed that MA is composed primarily of α-helices, which form a globular core that is connected to the CA domain by a C-terminal extended helix. 32 –34 The MA domain is cotranslationally modified at its N-terminus with myristate; as is the case with many other myristylated proteins, this covalent modification is essential for the association of HIV-1 Gag with the inner leaflet of the plasma membrane. Mutations that prevent Gag myristylation have a profound effect on virus assembly and release by blocking the association of Gag with the cell membrane. 35 –38 Resh and colleagues proposed that Gag–membrane binding is regulated by a “myristyl switch” mechanism, whereby the myristate can shift between exposed and sequestered conformations. 39 Summers and co-workers used nuclear magnetic resonance (NMR) methods to confirm this hypothesis and to demonstrate that myristate exposure can be promoted by factors that increase Gag self-association. 40 Mutations near the N-terminus of MA that disrupt Gag–membrane binding 41 apparently do so by preventing myristate exposure. 42
HIV-1 assembly takes place in cholesterol- and sphingolipid-enriched plasma membrane microdomains known as lipid rafts. 16,17,43 HIV-1 virions are thus highly enriched in cholesterol and saturated, raft-derived lipids. 44 –46 Gag displays behavior characteristic of a raft-associated protein in biochemical assays, 47 –51 cholesterol depletion of virus-producing cells inhibits virus particle production and virion infectivity, 49 and treating Gag-expressing cells with unsaturated fatty acids reduces virion assembly by directing Gag to nonraft domains on the plasma membrane. 52
In addition to the N-terminal myristate, Gag association with membrane is also regulated by a highly basic patch of amino acid residues located near the N-terminus of the MA domain (amino acid residues ∼17–31). 53,54 This basic cluster of amino acids forms a positively charged surface at the “top” of the MA domain. 32,33 Mutations in these basic residues mistarget Gag to late endosomes or multivesicular bodies (MVBs). 55 A number of cellular proteins whose membrane association is regulated by fatty acylation and a basic patch of amino acid residues interact with a specific class of negatively charged lipids known as phosphoinositides. The phosphoinositides differ from one another in the position and number of phosphates on the inositol head group. 56,57
Specific members of the phophoinositide family are enriched at particular membrane sites within the cell and thus their binding to proteins plays important roles in membrane and protein trafficking and signaling. 58 For example, phosphatidylinositol-(4,5)-bisphosphate [PI(4,5)P2] is enriched in the inner leaflet of the plasma membrane 57 ; phosphatidylinositol-(3,5)-bisphosphate [PI(3,5)P2] is concentrated on late endosomes 58 and phosphatidylinositol-3-phosphate [PI(3)P] is most abundant on early endosomal membranes and on late endosomal vesicles. 58 PI(4,5)P2 plays an important role in the association of HIV-1 Gag with the plasma membrane. 59 Overexpression of 5-phosphatase IV, an enzyme that converts PI(4,5)P2 to PI(4)P, results in the depletion of PI(4,5)P2 from the plasma membrane and induces the retargeting of Gag from the plasma membrane to late endosomes/MVBs. Overexpressing a constitutively active mutant of ADP-ribosylation factor 6 (Arf6) leads to the formation of PI(4,5)P2-enriched vesicular structures that serve as sites for Gag assembly. 59 These observations suggest that PI(4,5)P2 in the plasma membrane is required for efficient HIV-1 release, and that basic residues in MA are involved in direct interactions with PI(4,5)P2.
A direct interaction between MA and PI(4,5)P2 has been demonstrated by several groups using different approaches. Summers and co-workers used NMR spectroscopy to resolve the structure of a complex between a water-soluble, truncated form of PI(4,5)P2 and myristylated MA. 60 In this study, it was shown that MA binding to PI(4,5)P2 triggered the myristyl switch, resulting in increased myristate exposure. The NMR data revealed that in addition to electrostatic interactions between MA and PI(4,5)P2, the 2′-unsaturated acyl chain of PI(4,5)P2 packs into a hydrophobic cleft within the globular core of MA (Fig. 3).
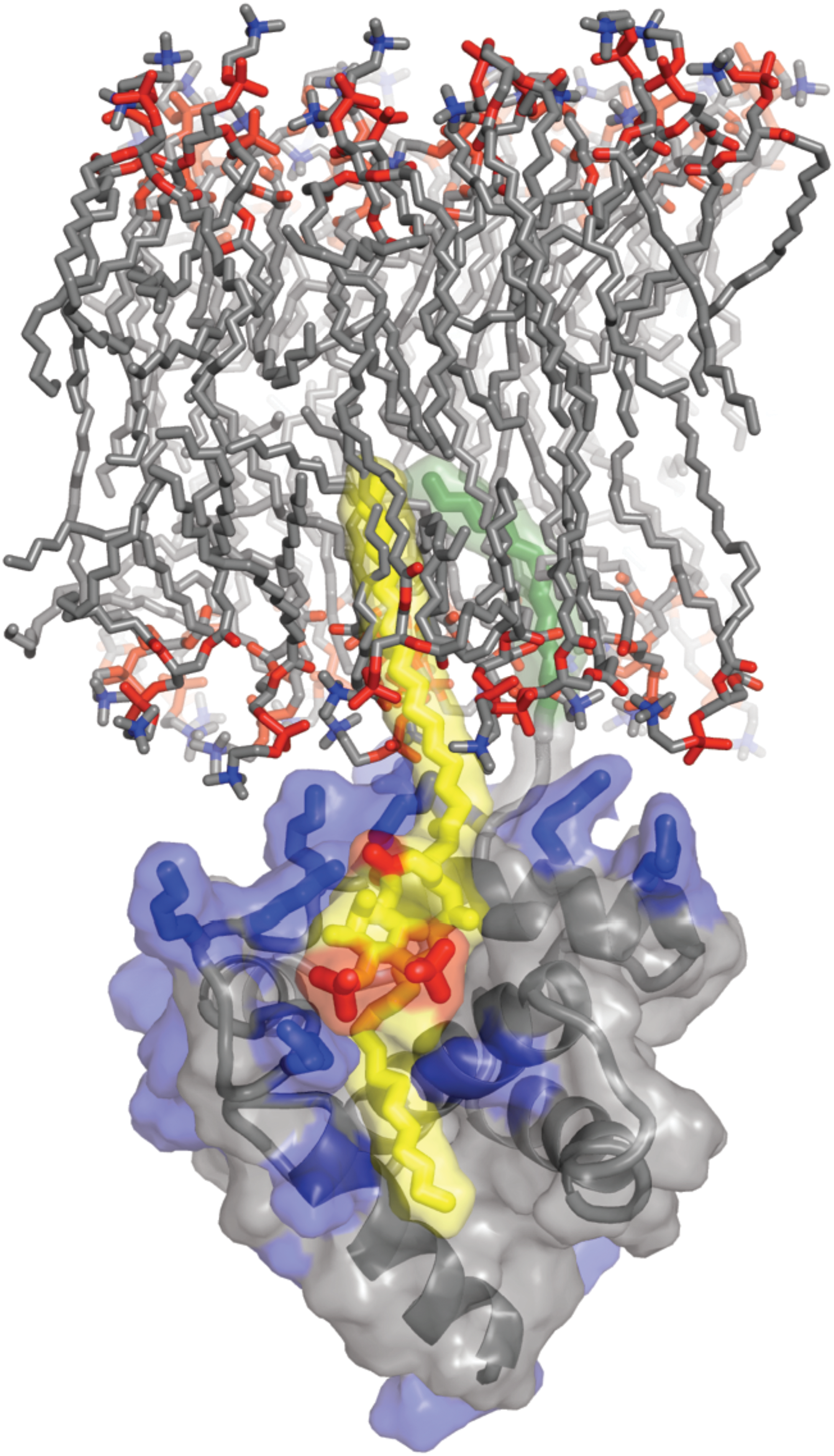
Model of HIV-1 MA binding to phosphatidylinositol-4,5-bisphosphate [PI(4,5)P2] in the membrane. The basic residues of MA (blue) interact electrostatically with phosphate groups (red) in PI(4,5)P2 (yellow and red). PI(4,5)P2 binding by MA induces exposure of the myristyl group (green), allowing it to insert into the membrane. The 2′-unsaturated acyl chain (yellow) of PI(4,5)P2 is predicted to bind to a hydrophobic cleft in MA. Unpublished image provided by M. Summers, based on the data of Saad et al. (2006).
60
Color images available online at
The extrusion of the unsaturated 2′-acyl chain of PI(4,5)P2 from the lipid bilayer upon MA binding could in theory promote Gag partitioning into the saturated lipid raft microenvironment, as the saturated 1′-acyl chain would be the sole PI(4,5)P2 acyl chain left in the membrane. Whether such a conformation would be energetically favorable in the context of a lipid bilayer remains to be determined. Shkriabai et al. used a mass spectrometric protein footprinting approach with lysine-modifying agents to show that Lys-29 and Lys-31 of MA are important for the MA–PI(4,5)P2 interaction. 61
These two basic residues are among those previously shown to be important for Gag localization and virus assembly. 55,62,63 Ono and co-workers used liposome-binding assays to show that full-length myristylated Gag binds to PI(4,5)P2-enriched vesicles, and that mutation of residues 29 and 31 reduced binding. 64 As one would predict if Gag–membrane association and HIV-1 assembly require PI(4,5)P2, this lipid is enriched in HIV-1 virions compared to the plasma membrane of the virus-producing cell. Furthermore, deletion of MA significantly reduced the incorporation of PI(4,5)P2 into virions. 46 Direct interactions between MA and PI(4,5)P2 have also been reported for other retroviruses, e.g., HIV-2, 65 MLV, 46,66 and EIAV. 67 These studies demonstrate that a broad array of retroviruses exploits an MA–PI(4,5)P2 interaction at the membrane binding step of virus assembly. In the case of EIAV, binding between MA and PI(3,5)P2 may regulate Gag association with internal membranes. 68
A number of host cell proteins have been implicated in regulating the trafficking of Gag to the plasma membrane. These include the clathrin adaptor protein complexes AP-1, AP-2, and AP-3, 69 –71 the Arf proteins, 72,73 the Golgi-localized, γ-ear-containing, Arf-binding (GGA) proteins, 72,73 tail-interacting protein 47 (TIP47), 74 –76 calmodulin, 77,78 and the soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE) machinery. 79 The mechanism by which these factors promote Gag–membrane association remains to be defined.
In addition to playing an important role in directing HIV-1 Gag to the plasma membrane, the MA domain also functions to direct the incorporation of the Env glycoprotein complex into virus particles. 80 While solid and reproducible biochemical evidence for a direct MA–gp41 interaction is lacking, abundant genetic evidence supports the existence of such an interaction. 62,81 –84 Host factors, including calmodulin and TIP47, have been implicated as cofactors in the Env incorporation process, 76,85 although a role for these proteins awaits independent confirmation and mechanistic characterization.
In theory, a variety of strategies could be pursued for therapeutically targeting the MA domain of HIV-1 Gag. As mentioned above, in its sequestered state, the myristate moiety attached to the N-terminus of MA packs into a hydrophobic groove in the globular core of MA. 40,60 This hydrophobic cleft could potentially be targeted by small molecules whose binding would dysregulate the myristyl switch. This could result in enhanced Gag–membrane association, which intuitively would seem likely to increase the efficiency of virus assembly; however, mutations that enhance membrane association have been reported to disrupt replication by inducing a postentry defect in the next round of infection. 86 The PI(4,5)P2-binding cleft of MA could also be targeted by small molecules that would compete for an MA–PI(4,5)P2 association. Interestingly, in vitro studies have found that soluble PI(4,5)P2 derivatives, rather than competing with MA for PI(4,5)P2 binding and thus disrupting membrane association, actually enhance the efficiency of MA binding to liposomes. 87
This raises the possibility that small molecules that dock in the PI(4,5)P2-binding groove might increase Gag–membrane association by triggering the myristyl switch. Such dysregulation of membrane binding activity could disrupt proper Gag targeting, or, as suggested above, could interfere with particle infectivity. If the putative interaction site between MA and the gp41 Env glycoprotein can be better defined, the MA–gp41 interaction could possibly be targeted to disrupt Env incorporation into virions. The association of Gag with lipid rafts could also be developed as a therapeutic target, as reviewed in detail recently, 16,17 perhaps most effectively in the context of chemoprevention. Finally, the interaction between MA and host proteins could be disrupted pharmacologically.
Several issues need to be addressed before significant progress can be made in developing antiretrovirals that target the MA domain: (1) robust, high-throughput assays, preferably cell-based, need to be developed to screen for molecules that block the association of Gag with membrane and/or disrupt the proper localization of Gag at the plasma membrane, and (2) structural information needs to be obtained on putative interaction interfaces between MA and gp41 and between MA and host-cell factors. With this additional information and related screening tools, the MA domain could offer a variety of possibilities for antiretroviral development.
CA
As is the case with MA, the CA domain and the mature CA protein play multiple roles in the virus replication cycle. During assembly, the CA domain of Pr55Gag functions to promote Gag–Gag interactions that drive Gag multimerization; after release and cleavage of Pr55Gag by the viral PR, the mature CA protein plays a central role in particle maturation by reassembling into the conical core that houses the viral RNA genome and the viral enzymes RT and IN. 88,89 After entry into the target cell, the CA core complex serves as the target for host restriction factors (e.g., TRIM5α). CA is composed of two independently folded domains, the N-terminal and C-terminal domains (NTD and CTD, respectively). The CA NTD and CTD are connected by a short flexible linker. 89,90 The NTD (CA residues 1–145) is composed of seven α-helices (CA helices 1–7) packed in the shape of an arrowhead, with an extended, Pro-rich loop connecting helices 4 and 5. This loop binds the peptidyl-prolyl cis-trans isomerase, cyclophilin A. 91 –93 Deletion of the entire NTD does not disrupt particle production; however, point mutations in helices 4–6 of the NTD impair particle production, suggesting that this region of the NTD forms weak interactions during assembly or that these NTD mutations disrupt overall CA folding. 94 –96
The CTD (residues 151–231) is composed of a short 310-helix followed by an extended strand and four α-helices (CA helices 8–11). The CA CTD has a propensity to dimerize 97,98 and plays a central role in Gag multimerization during assembly. 94 –99 CA dimerization takes place by mutual interactions of α-helix 9 from each monomer, with the aromatic rings of Trp-184 buried in the dimer interface. 97,98 Mutation of residues 184 and 185 significantly reduces particle production in cells and CA dimerization in vitro. 96,97,100,101 Near the N-terminus of the CTD, in helix 8, is a sequence of 20 amino acids that is highly conserved among retroviral CA proteins. 97,102,103 This region, aptly named the major homology region (MHR), plays an important role in Gag assembly. 96,102,104,105 The crystal structure of the CTD shows that the MHR is distinct from the dimer interface and forms an array of hydrogen bonds. 97 The precise role of the MHR in CA function remains to be fully elucidated.
Residues at the C-terminus of CA and the N-terminus of SP1 are also important for immature particle formation, suggesting that the CA/SP1 boundary region forms a critical assembly domain. 94,96,106 –112 Although this region is highly flexible and disordered in crystal structures, it displays a tendency to form an α-helix. 31,94,97,98,113,114 It has been suggested from cryoelectron tomography analysis that the putative CA/SP1 boundary region forms a six-helix bundle. 31
In the immature particle, the CA domain assembles into a hexameric lattice, with a unit-to-unit spacing of ∼8 nm. 25,115 The immature hexameric lattice is continuous but incomplete, with a large gap that allows the lattice to curve. 30 During PR-mediated maturation, ∼1500 copies of CA again assemble into a hexagonal lattice, but with a unit cell spacing of ∼10 nm. 116 In its mature form, the hexagonal lattice organization resembles that of a fullerene cone, with the hexameric tubes closed off at both ends by the presence of a specific number (12) of pentamers 116,117 (Fig. 4). The hexameric lattice of the mature conical core is composed of rings formed by six NTDs connected to each other by CTDs (Fig. 4). Several stabilizing interfaces create the hexameric rings and link them together to form the lattice: (1) NTD–NTD intermolecular interactions create the NTD hexameric rings, (2) NTD–CTD intermolecular interactions stabilize the hexamer, and (3) the CTD–CTD dimer contacts link the rings together to form the lattice. 118 –120 Recently, Gronenborn and colleagues described an additional CTD–CTD interface formed by CTD dimers from three adjacent hexamers. 121,122 Only relatively subtle differences are observed between the subunit interfaces of pentamers and hexamers. 123,124
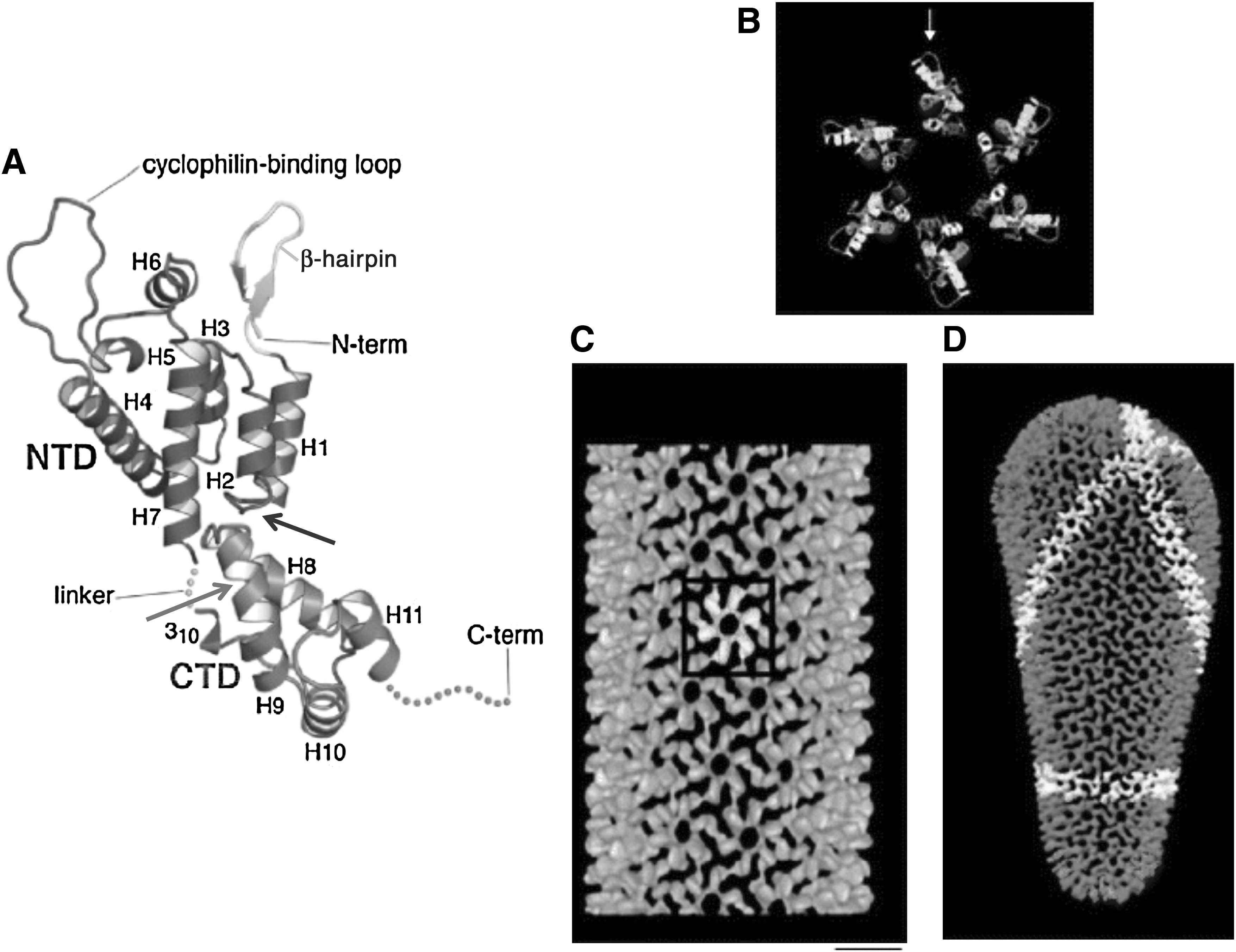
Structure of mature HIV-1 CA.
The detailed structural information now available on HIV-1 CA, obtained over the past dozen years by a combination of cryoelectron tomography, NMR, and x-ray crystallography methods, backed up by extensive mutations analyses, offers a diversity of targets for inhibitor development. In principle, inhibitors that bind to CA could potentially disrupt the assembly of both immature viral particles and mature cores. In initial studies, isolated CTDs were used as competitive inhibitors for CA assembly in vitro. 125,126 Small peptides or organic molecules that bind at or near the CTD dimerization interface hold the potential to weaken CA dimerization. An early attempt was made by Hilpert et al. using sequence-derived peptides that inhibited the dimerization of CA in solution. 127 The peptide IPVGEIYKRW, corresponding to α-helix 7 of the NTD, was reported to suppress CA dimerization in solution. However, the mechanism responsible for this inhibition remains to be clarified, and whether this peptide displays antiviral activity in cell-based systems has not, to our knowledge, been reported. Neira and co-workers designed a synthetic peptide termed CAC1 that is based on the sequence of CA helix 9, and showed that it binds to the CTD in solution. 128
An in silico screening approach was used by Summers and colleagues to identify a small organic molecule, N-(3-chloro-4-methylphenyl)-N′-[2-[([5-[(dimethylamino)- methyl]-2-furyl]-methyl)-sulfanyl]ethyl]urea (CAP-1) that binds to the CA NTD. 129 As a consequence of this interaction, virus assembly and maturation are disrupted in cell-based assays. NMR and x-ray crystallographic studies mapped the binding site of CAP-1 to a pocket formed at the point at which helices 1, 2, 4, and 7 of the NTD interact 130 (Fig. 5). Binding of CAP-1 to the NTD induces the displacement of the deeply buried aromatic ring of Phe-32, thereby creating the CAP-1 binding site. 130 Thus, the CAP-1 interaction with the CTD is largely hydrophobic and appears to disrupt the docking of the NTD and CTD. Although CAP-1 displays relatively weak binding affinity for CA and significant cytotoxicity, the Phe-32 pocket will likely serve as a useful target for future CA-based drug development.
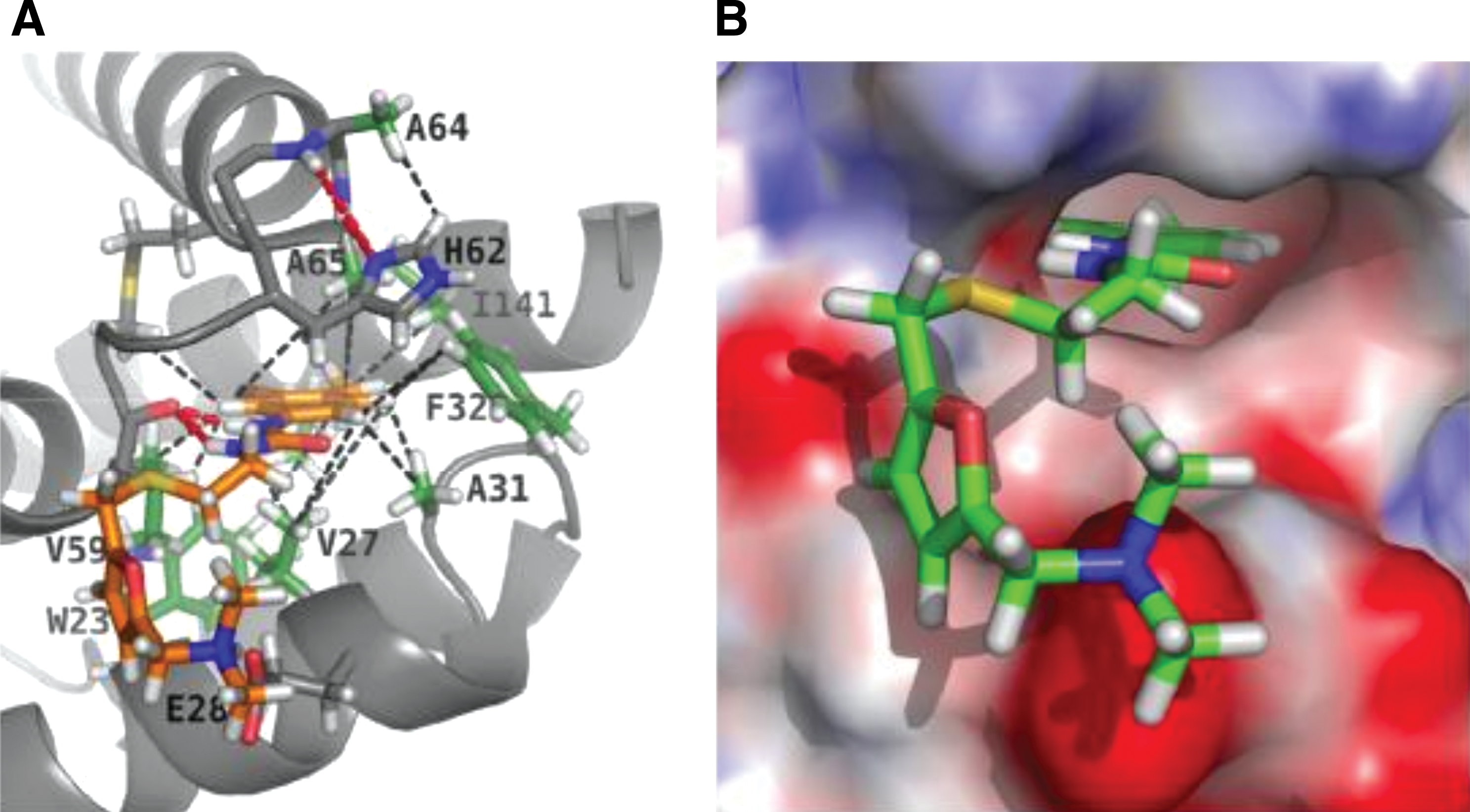
Structure of CAP-1 bound to the NTD of CA calculated by restrained molecular dynamics based on hybrid x-ray/NMR data.
Krausslich and co-workers used a phage display screen with both full-length CA and the CTD-SP1-NC region of Gag as bait to identify a CTD-binding peptide termed CAI (CA assembly inhibitor). 131 The sequence of CAI, ITFEDLLDYYGP, is not homologous to the dimerizing domain of the CTD but inhibits the in vitro assembly of both spherical particles that are analogous to immature VLPs and tubular structures that possess a mature-like CA organization. 131 A high-resolution x-ray structure of the CAI–CTD complex reveals that the peptide binds in an α-helical conformation to a hydrophobic groove between helices 1, 2, and 4 132 (Fig. 6). The binding of CAI allosterically disrupts the CA dimer interface and interferes with contacts between the CTD and the NTD. Barklis et al. reported that CAI induces the disassembly of CA tubes by modifying the CTD interface and also replaces NTD α-helix 4 in its binding to a CTD groove that helps to align NTDs and CTDs around the CA hexamers. 133 Thus, the binding of CAI to the CTD weakens the hexamer, thereby impairing CA assembly and potentially destabilizing the viral core.

Structure of the CAI peptide bound to a hydrophobic cavity of the CA CTD. The surface of the CTD is colored based on polarity: red, positively charged; blue, negatively charged; cyan, uncharged; white, nonpolar. The residues of CAI (yellow) in direct contact with the CTD, and N- and C-termini of CAI, are labeled. Reprinted from Ternois et al. (2005)
132
with permission. Copyright 2005 Macmillan Publishers Ltd. [Nature Structural and Molecular Biology]. Color images available online at
CAI itself does not display antiviral activity because it is not cell permeable; this limitation, however, was addressed by using a technique known as hydrocarbon stapling to generate cyclical, cell-penetrating derivatives with increased α-helical character. 134,135 The resulting peptides, NYAD-1 and its water-soluble derivative NYAD-13, contain a covalent bridge between two amino acids separated by four residues. The sequence of NYAD-1 is ITFXDLLXYYGKKK, where X is the nonstandard amino acid (S)-2-(2′-pentenyl) alanine, which helps to stabilize the peptide. These peptides penetrate into cells and disrupt the assembly of immature VLPs, inhibit virus maturation, and exhibit an antiviral effect. 135 NMR chemical shift perturbation studies indicate that CAI and NYAD-1 bind to the same hydrophobic pocket in the CTD. 134,135 This hydrophobic pocket was subjected to an in silico docking-based screen for small molecules that could potentially interfere with CA CTD function. Several molecules that disrupted in vitro CA assembly and virus infectivity resulted from this effort. 136
Debnath and co-workers also designed a dimer interface peptide, NYAD-201, containing the sequence AQEVKXWMTXTLLVA [based on the sequence of CA amino acids 178–192, where X represents the (S)-2-(2′-pentenyl) alanines that are connected by hydrocarbon stapling 137 ]. This stapled peptide penetrates into cells and inhibits HIV-1 production by 3-fold. This inhibition in virus production is specific to NYAD-201, as a mutant peptide derivative in which the key dimer interface residues “WM” were changed to “AA” does not inhibit virus release. In vitro CA assembly is disrupted by NYAD-201 but not the mutant control peptide. In addition to inhibiting virus production, NYAD-201 also disrupts HIV-1 infectivity in an Env-dependent manner. 137 NYAD-201 inhibits replication of a large panel of laboratory-adapted and primary HIV-1 isolates. 137
As mentioned above, a Pro-rich loop in the CA NTD binds the peptidyl-prolyl isomerase cyclophilin A. Early studies demonstrated that disruption of the CA–cyclophilin A interaction with cyclosporine inhibited HIV-1 replication. 138,139 More recently, a nonimmunosuppressive analog of cyclosporine, Debio-025, was shown to inhibit an early step in HIV-1 replication with increased potency and reduced cytotoxicity relative to cyclosporine. 140
In a high-throughput screen for compounds that inhibit HIV-1 replication, researchers at Pfizer identified a compound, PF-1385801, that targets the CA NTD. 141 Blair et al. prepared several analogs of this compound, two of which—PF-3450074 and PF-3759857—were active against most HIV-1 isolates with an effective concentration <1 μM. 142 Interestingly, these compounds inhibited both early and late stages of HIV-1 replication. The early block was imposed after viral entry but before reverse transcription, and the late block was at the level of virus assembly. 142 A high-resolution cocrystal structure of CA and PF-3450074 revealed that the compound binds to the pocket created by helices 3, 4, 5, and 7 of the CA NTD. Selections in PF-3450074 led to substitutions in CA that conferred resistance to the compound. 142 In vitro experiments using HIV-1 particles or purified viral cores showed that PF-3450074 destabilizes the viral core, and in target cells, PF-3450074 appears to inhibit HIV-1 infection by triggering premature uncoating. 143
Researchers at Boehringer Ingelheim converted the CA–NC in vitro assembly assay 144 into a high-throughput format and screened for inhibitors of in vitro assembly. Two distinct classes of compounds were identified, benzodiazepines and benzimidazoles. 145 Both classes of compounds appear to bind the CAP-1 pocket discussed above, and disrupt particle formation and virion maturation. 145 Optimization of the benzodiazepine series led to a compound with significantly improved potency. 146 Together, these studies indicate that HIV-1 CA is a potential drug target.
NC
The NC domain of HIV-1 Gag is characterized by the presence of two zinc finger-like motifs flanked by highly basic sequences (Fig. 7). 147,148 NC zinc fingers, typically present in one or two copies, are among the most highly conserved features of retroviral Gag proteins, and, as could be predicted from this high level of conservation, are critical for NC function. The consensus zinc-finger motif in retroviral NC domains is Cys-X2-Cys-X4-His-X4-Cys (CCHC), a sequence that is somewhat rare among cellular zinc fingers. As a domain of Pr55Gag, NC serves several primary functions during the assembly process: it enhances Gag–Gag interactions, largely as a consequence of its nucleic acid binding properties; it directs the packaging of the viral RNA genome into virus particles; and it promotes the incorporation into virions of tRNALys3, which serves as the primer for reverse transcription in the next round of infection. 7 In general, the basic residues in NC are responsible for the Gag multimerization activity, again, due to their nonspecific binding to nucleic acid. Gag-bound nucleic acid serves as a multimerization platform, or scaffold, that serves to concentrate Gag molecules and facilitate their assembly.
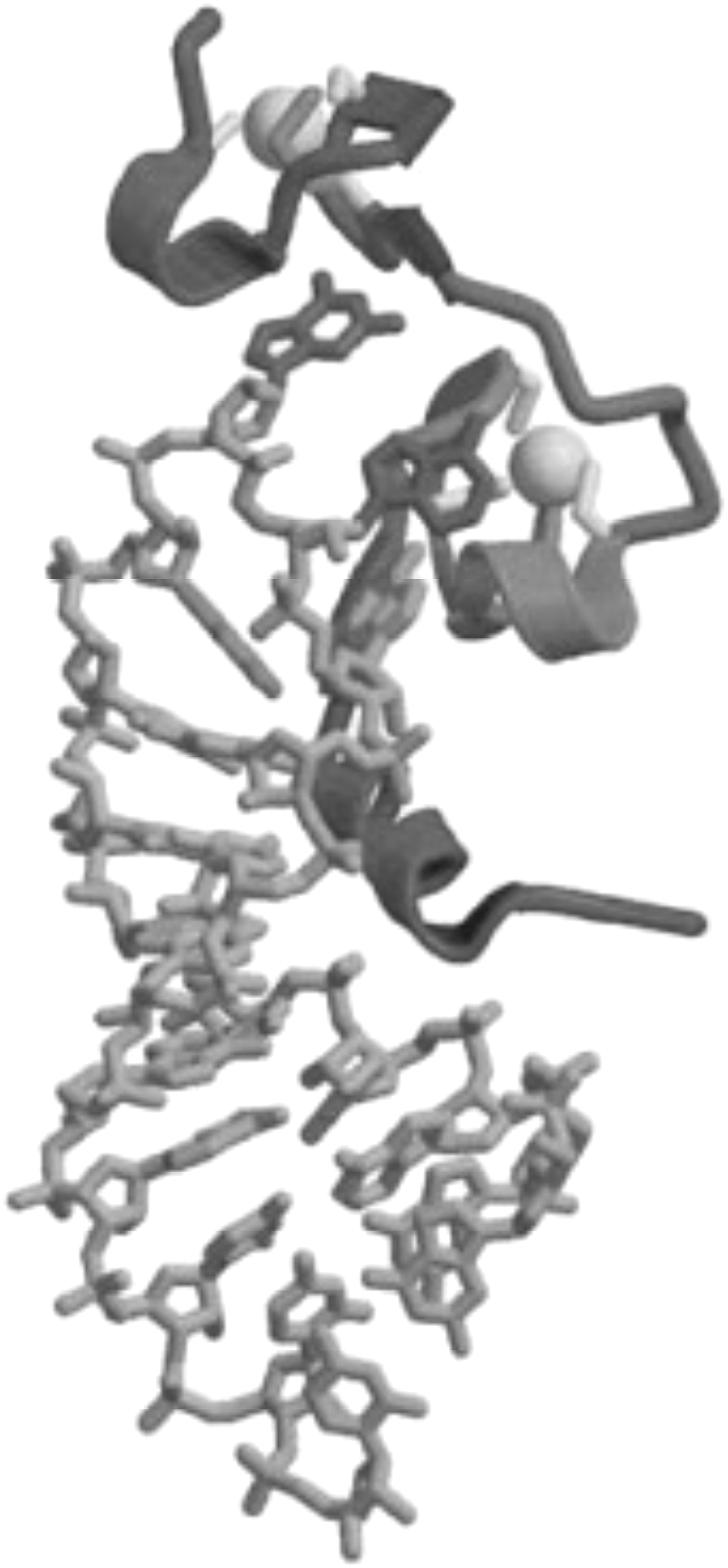
Structure of NC bound to the SL3 stem-loop of the HIV-1 RNA packaging signal. Gray balls represent the two zinc ions that bound to the zinc-finger motifs. Reprinted from Turner and Summers (1999) 278 with permission. Copyright 1999 Elsevier Inc.
In contrast, specific viral genomic RNA encapsidation involves the interaction between the NC zinc fingers and a multiple stem-loop structure in the viral RNA known as the packaging, or ψ, site. 149 –163 Gag selects and packages dimeric viral RNA from the cytosol. 164 After liberation from Pr55Gag following PR-mediated Gag processing, the mature 7-kDa NC protein functions as a nucleic acid chaperone to increase the efficiency of reverse transcription and integration. 7
The critical roles of NC in HIV-1 replication, together with the high degree of conservation and unusual sequence motif of the NC zinc fingers, suggest that NC should be a good target for drug development. Over the past nearly two decades, a number of NC-targeted HIV-1 inhibitors have been reported, most of which act by disrupting the zinc fingers. Rice and colleagues reported that cell-permeable compounds that oxidize the NC zinc-finger cysteines, thereby destroying the zinc-finger structure, inactivate virus infectivity. 165,166
One such compound, 2,2-dipyridyl disulfide (also known as Aldrithiol-2 or AT-2), induces extensive cross-linking within NC and Pr55Gag and is frequently used as a research tool to chemically inactivate HIV-1. 167 –169 The alkylating agent N-ethylmaleimide (NEM), attacks the zinc-bound thiols in NC and consequently impairs virus infectivity. 170,171 Appella and co-workers reported that pyridinioalkanoyl thioesters (PATEs) that are based on a 2-mercaptobenzamide thioester chemotype selectively target the zinc fingers of NC without affecting the zinc fingers found in other viral or cellular proteins. 172 –174 These PATE compounds mediate zinc ejection from NC and display low-micromolar antiviral activity with low cytotoxicity. 173,174
Interestingly, one of these compounds is reportedly efficacious in an HIV-1 transgenic mouse model system to suppress the production of infectious virus particles. 175,176 These initial studies led to the development of a new class of zinc-ejecting compounds, the N-substituted S-acyl-2-mercaptobenzamide thioesters (SAMTs). 177,178 Unlike other NC inhibitors, the SAMTs use an S-acyl transfer mechanism rather than an oxidative mechanism to eject zinc. 177,179 These compounds were reported to show antiviral activity as a vaginal microbicide in rhesus macaques in an SIV/HIV-1 (SHIV) chimeric virus infection model. 180 SAMTs possess an interesting mechanism of action; they interact with NC and covalently modify the protein with an acetyl group, thereby inhibiting Gag processing and viral infectivity. 181
Reportedly, the resulting mercaptobenzamide thiol can be reacetylated in cells with acetyl-CoA to regenerate the reactive thioester form of the inhibitor, which can then react with another molecule of Gag. 181 This recycling mechanism could potentially increase the potency of the SAMT class of inhibitors. In another recent study, Gorelick and co-workers showed that a sulfhydryl compound [4-vinylpyridine (4-VP)] in conjunction with a membrane-permeable divalent metal ion chelator, binds to cysteine thiols in the NC zinc fingers and inactivates HIV-1 infectivity. 182
The compounds described above act principally by attacking the zinc ions bound in the NC zinc fingers. The nucleic acid chaperone activity of NC could also be targeted. To this end, de Rocquigny and colleagues developed a screen for molecules that inhibit the nucleic acid destabilizing activity of NC. 183 This assay is based on the ability of NC to unfold a DNA stem-loop that is labeled on one end with a fluorophore and on the other end with a fluorescence quencher. Five compounds with IC50 values in the micromolar range were identified that bind to NC zinc fingers and prevent the protein from interacting with the DNA stem-loop. These molecules block binding of NC to DNA without promoting zinc ejection. 183 An alternative approach to disrupting NC function targets RNA encapsidation. Along these lines, Turner and colleagues reported that aminoglycosidic antibiotics were capable of interfering with genome recognition and encapsidation. 184 The steady progress in developing ever-more-potent NC-based inhibitors suggests that this area will remain active in the future.
p6
The p6 domain, located at the C-terminus of HIV-1 Gag, functions late in the virus assembly pathway to promote the pinching off of virus particles from the cell surface. An early study observed that deletion of the p6 domain from HIV-1 Gag produced a severe block to particle production. 185 Intriguingly, EM analysis showed that the p6-deleted particles were fully assembled but failed to bud off from the plasma membrane, instead remaining attached to the cell surface by a thin tether. Subsequent work identified a highly conserved Pro-Thr/Ser-Ala-Pro [P(T/S)AP] motif as being responsible for the budding activity of p6186; mutation of any of these residues recapitulated the phenotype observed upon p6 deletion. 186,187 Domains with analogous functions were identified in the Gag proteins of a number of other retroviruses. These motifs were named “late domains” because of their role in virus budding. 188 Three classes of late domains have been identified in retroviral Gag proteins: PT/SAP, PPXY, and YP(X) n L (where “X” is any amino acid, and n=1–4 residues). 18 –20,189 In many cases, retroviral Gag proteins contain multiple late domains, often located within a few residues of each other.
In the case of HIV-1, for example, the primary late domain is PT/SAP, as mentioned above, but a secondary late domain of the YP(X) n L type is found near the C-terminus of p6. It should be noted that late domains bearing these sequences are also found in other nonretroviral enveloped viruses. For example, the matrix-like VP40 protein of Ebola virus contains overlapping PTAP and PPXY motifs. 190
A compelling body of evidence indicates that late domains act by interacting directly with host proteins that are part of the cellular endosomal sorting pathway. At the core of this machinery are several multiprotein complexes, originally identified in yeast by the laboratory of Scott Emr, that are known as endosomal sorting complexes required for transport (ESCRT) 0, I, II, and III (Fig. 8). 18,20,191,192 The ESCRT complexes play sequential roles in the sorting of cargo proteins into intralumenal vesicles that bud into late endosomes to generate MVBs. 191,193 The cargo-laden MVBs ultimately fuse with the lysosome, leading to cargo degradation. Sorting of cargo proteins into the MVB pathway often requires monoubiquitination of the cytoplasmic domains of the cargo and recognition of the ubiquitin moiety by the sorting apparatus. The ESCRT machinery also functions in the membrane scission reaction that occurs during cytokinesis. 194 –196 Retroviruses have thus evolved to usurp a cellular machinery that directs budding and membrane fission reactions that are oriented away from the cytosol, the topological equivalent of virus budding.
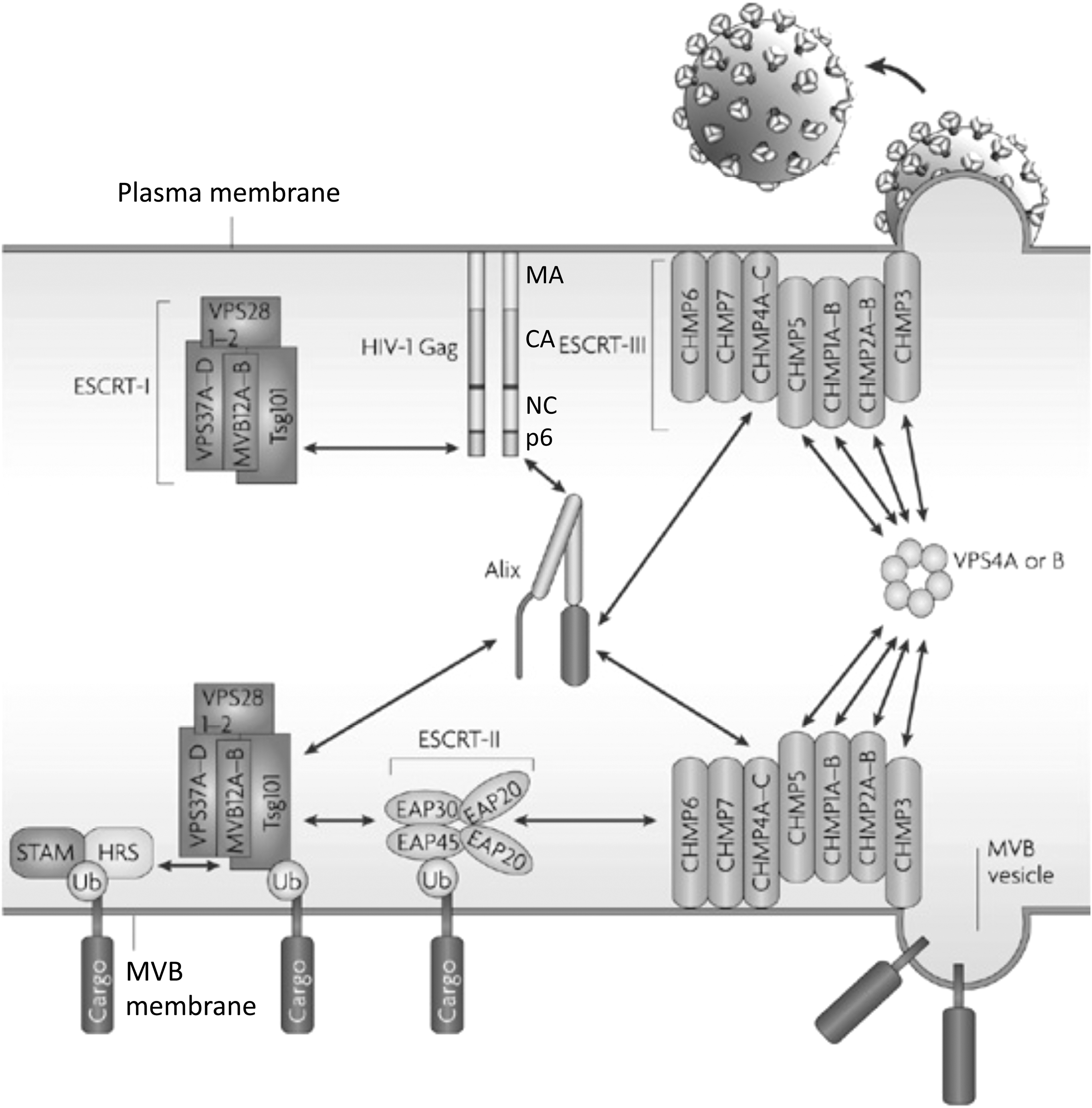
Schematic representation of the ESCRT machinery and its interaction with HIV-1 Gag at the plasma membrane and ubiquitinated cargo in the MVB. The MA, CA, NC, and p6 domains of Gag are shown. Double-headed arrows denote the interaction of p6 with Tsg101 and Alix, Alix with Tsg101, and Vps4 with ESCRT-III. A ubiquitinated (Ub) cargo protein is shown interacting with the STAM/Hrs complex (often referred to as ESCRT-0). Adapted with permission from Fujii et al. (2007). 279 Copyright 2007 Macmillan Publishers Ltd. (Nature Reviews Microbiology).
In addition to the four major ESCRT complexes—ESCRT-0, I, II, and III—a variety of ESCRT-accessory proteins participate in ESCRT function. These include Alix, a molecule that possesses interaction sites for both ESCRT-I and ESCRT-III, the AAA ATPase Vps4, and members of the Need4 family of ubiquitin ligases, which are involved in cargo ubiquitination. Retroviral late domains interact with various components of this sorting machinery. The PT/SAP motif binds the ubiquitin E2 variant (UEV) domain of the ESCRT-I component Tsg101 (Fig. 9), 197 –199 YP(X) n L motifs interact with Alix (Fig. 10), 200 –202 and PPPXY motifs bind Nedd4-family ubiquitin ligases. 18 –20
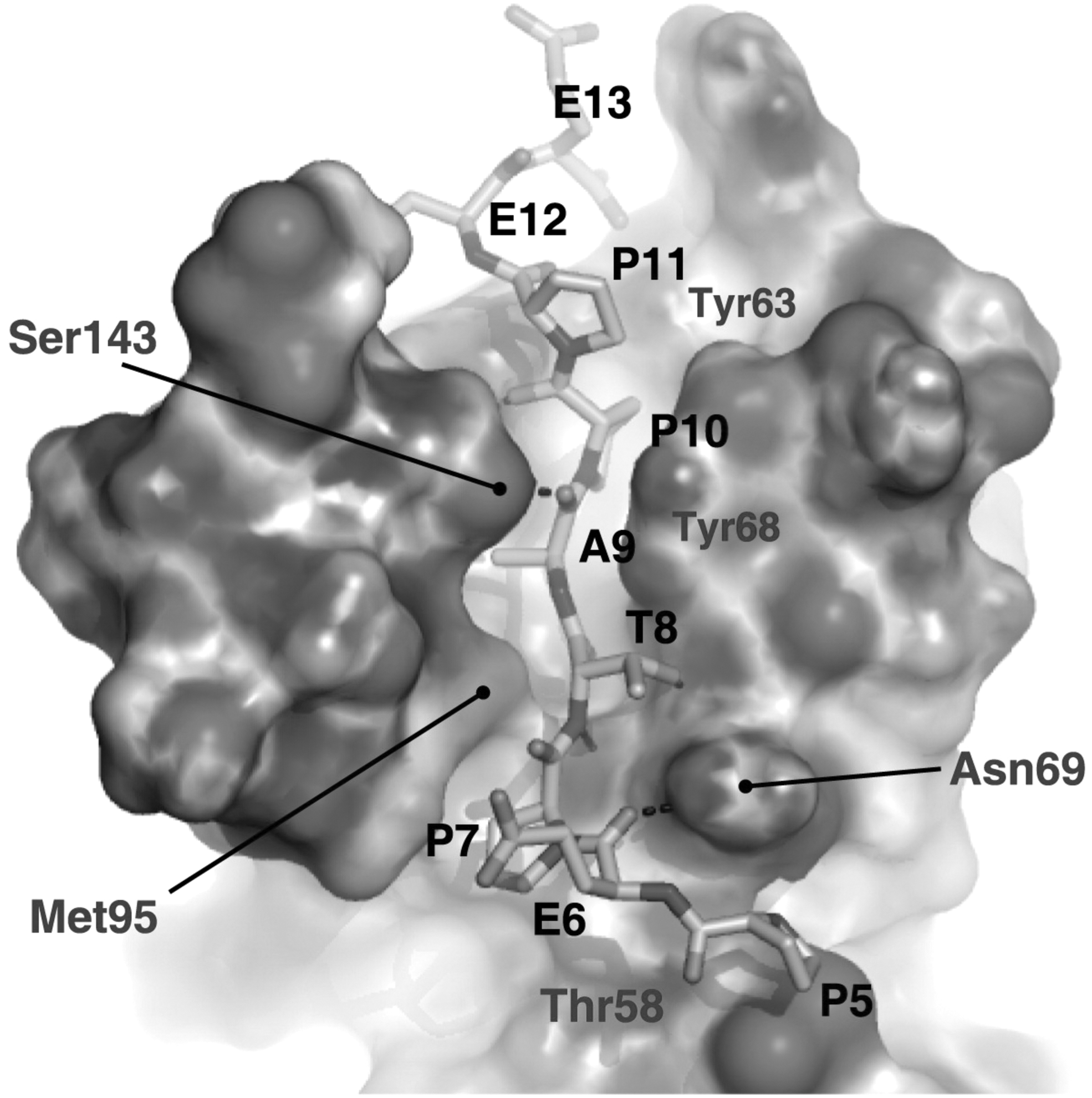
Structure of the UEV domain of Tsg101 bound to a PTAP-containing peptide derived from the sequence of HIV-1 p6. p6 residues Pro-5 (P5), Glu-6 (E6), Pro-7 (P7), Thr-8 (T8), Ala-9 (A9), Pro-10 (P10), Pro-11 (P11), Glu-12 (E12), and Glu-13 (E13) are shown, as are residues in Tsg101 that make contact with the PTAP-containing peptide. Reprinted with permission from Im et al. (2010). 211 Copyright 2010 Cell Press.
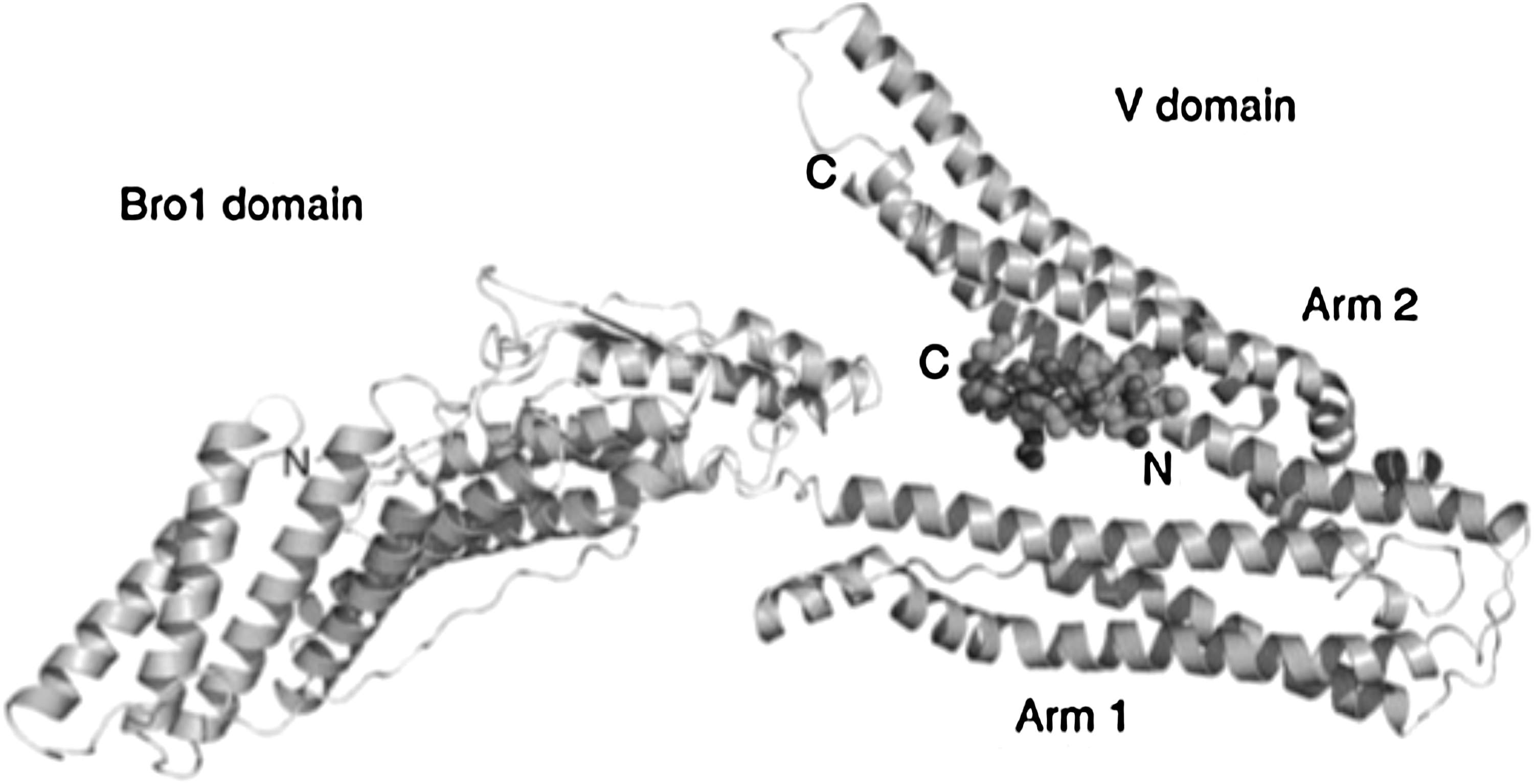
Structure of a YPXnL-containing peptide bound to the Alix V domain. Ribbon diagram of the Bro1 and V domain of ALIX in complex with the HIV-1 p6 YPXnL late-domain peptide. Adapted from Zhai et al. (2008). 210 Copyright 2007 Macmillan Publishers Ltd. (Nature Structural and Molecular Biology).
The functional significance of HIV-1 late domain interactions with ESCRT or associated machinery has been demonstrated by a number of studies showing that disruption of these interactions is detrimental to HIV-1 budding. As mentioned above, early studies indicated that mutation of the primary HIV-1 late domain, PT/SAP, led to the tethering of virions at the cell surface and failure of the virions to pinch off. 185,186 Subsequently, it was shown that Tsg101 depletion with siRNA or overexpression of the Gag-binding UEV domain of Tsg101 (referred to as TSG-5′) impaired virus release in much the same manner as PT/SAP mutation. 187,197 Fusion of Tsg101 to Gag can also stimulate the release of particles lacking the PT/SAP late domain. 203 Stable overexpression of TSG-5′ blocks the replication of feline immunodeficiency virus (FIV), which also encodes a Tsg101-binding, PT/SAP late domain. 204 Disruption of the Gag–Alix interaction potently inhibits EIAV release but produces more subtle effects on virus budding in the case of HIV-1. 200,201,205 –207 However, overexpression of the V-domain strongly disrupts HIV-1 budding, 205,206 and YP(X) n L mutations, although not producing large effects on virus release, do significantly delay HIV-1 replication in a range of cell types including primary lymphocytes and macrophages. 208
While overexpression of the Tsg101 UEV domain (TSG-5′) and the Alix-V domain inhibits HIV-1 budding, these large protein fragments are not viable as therapeutics. They do, however, provide proof-of-principle that Gag interactions with cellular endosomal sorting machinery constitute viable targets for developing antiviral inhibitors. Structural information obtained from NMR and x-ray crystallographic studies has provided high-resolution detail of the PT/SAP and YP(X) n L motif interactions with Tsg101 (Fig. 9) and Alix (Fig. 10), respectively. 198,209 –211 Binding affinities have also been calculated. These interactions are of relatively low affinity, consistent with the other protein–protein interactions along the ESCRT pathway. In an effort to design competitive inhibitors of the PT/SAP–Tsg101 interaction that display increased affinity for Tsg101, Burke and co-workers replaced the Pro residues in the peptide PEPTAPPEE with N-substituted glycines. This was accomplished by using hydrazone and hydrazide chemistry to synthesize a large series of these peptoid derivatives. 212
In a follow-up study, 213 each amino acid in the PEPTAPPEE peptide was replaced by amino-oxy-containing residues. These peptides were then reacted with a series of aldehydes to obtain a library of peptoids in which each residue was modified by different functional groups. Fluorescent anisotropy was used to evaluate the binding affinity of each peptoid for the UEV domain of Tsg101. In some cases, 15- to 20-fold increases in binding affinity were observed relative to the parental PEPTAPPEE peptide. 212,213 Finally, the binding conformations were stabilized and cellular uptake was increased by peptide circularization. 214 Efforts are underway in our laboratory to test these peptoids for their ability to inhibit HIV-1 budding in cell-based assays. In an independent study, Tavassoli et al. designed a bacterial reverse two-hybrid system (RTHS) to identify inhibitors of the p6–Tsg101 interaction. 215
They generated a large library (108) of cyclic peptides using the so-called SICLOPPS (split intein-mediated circular ligation of peptides and proteins) method, and these peptides were screened in RTHS to identify a small number of peptides that interfered with the p6–Tsg101 interaction. 215 After several rounds of secondary screening, four cyclic peptides were identified that inhibited these interactions, and one of these peptides (IYWNVSGW) showed a modest ability (∼2-fold) to inhibit the release of virus-like particles in cell culture. 215 The inhibitory activity of the peptide is reportedly PT/SAP-dependent, as release of an HIV-1 PTAP mutant was not inhibited. These studies represent initial steps in developing inhibitors of the interaction between p6 and Tsg101. Additional studies are required to identify small molecules capable of disrupting HIV-1 budding.
Thus far, efforts have focused on the PT/SAP–Tsg101 interface, as this is the primary late-domain interaction required for HIV-1 budding. Whether effective disruption of HIV-1 release will also require blocking the p6–Alix interaction remains to be tested. The observation that PT/SAP and YP(X) n L mutations synergize to completely block HIV-1 replication 208 suggests that this might be the case. Also unresolved is whether targeting Gag interactions with the ESCRT machinery will also block vital interactions between cellular proteins and thus result in significant cytotoxicity. Efforts are currently underway in our laboratory to develop small molecule inhibitors of the PT/SAP–Tsg101 interaction.
Maturation
As discussed above, HIV-1 maturation is triggered by the cleavage of the Gag polyprotein Pr55Gag by the viral PR. The proteolytic processing of Pr55Gag takes place in a sequential cascade of events: first, Gag is cleaved into two fragments, MA–CA–SP1 and NC–SP2–p6; then cleavage occurs at the MA–CA and SP2–p6 junctions; finally, CA–SP1 and NC–SP2 are cleaved to liberate CA and NC domains. 216 –222 The newly generated mature CA assembles to form the conical core, a process that is essential for virus infectivity. 25,223 Processing of Gag by PR is also required for activation of the fusogenic activity of the viral Env glycoprotein. 83,224 Mature NC is involved in stabilization of viral dimeric RNA and condensation of the viral ribonucleoprotein complex. 225,226
One of the important features of Gag processing is that it occurs in a highly ordered sequence, with the rate of processing at each step kinetically regulated in part by widely differing affinities of the PR for each of the individual cleavage sites. It is well established that interfering with the cleavage at any of the sites in Gag, or changing the order with which the sites are cleaved, results in aberrant virion morphology and loss of infectivity. 94,108,219,222,227 –229 These observations suggest that disrupting Gag processing event(s) could be a viable pharmacological approach for inhibiting HIV-1 replication. Although PR inhibitors (PIs) that inhibit the enzymatic activity of the enzyme and block Gag and Gag-Pol processing have been used effectively for many years, an alternative approach is to target individual Gag cleavage sites. Thus, inhibitors that block maturation by targeting Gag rather than PR are known as “maturation inhibitors” and are distinct from PIs. 21,89
Although in theory inhibitors could be developed that block each of the five Gag cleavage sites, until now only the CA–SP1 cleavage site has been successfully targeted. The small molecule 3-O-(3′,3′-dimethylsuccinyl)betulinic acid (DSB), also known as PA-457 or bevirimat (BVM) (Fig. 11), is the first member of this class of inhibitors. 229,230 BVM disrupts a late stage in PR-mediated Gag processing by blocking the cleavage of SP1 from CA, leading to the accumulation of the Gag processing intermediate CA–SP1. 229,230 The accumulation of CA–SP1 interferes with virion maturation, leading to the formation of particles that lack conical cores but instead display an acentric aggregation of electron density and a crescent of Gag density near the viral membrane (Fig. 11). 230,231
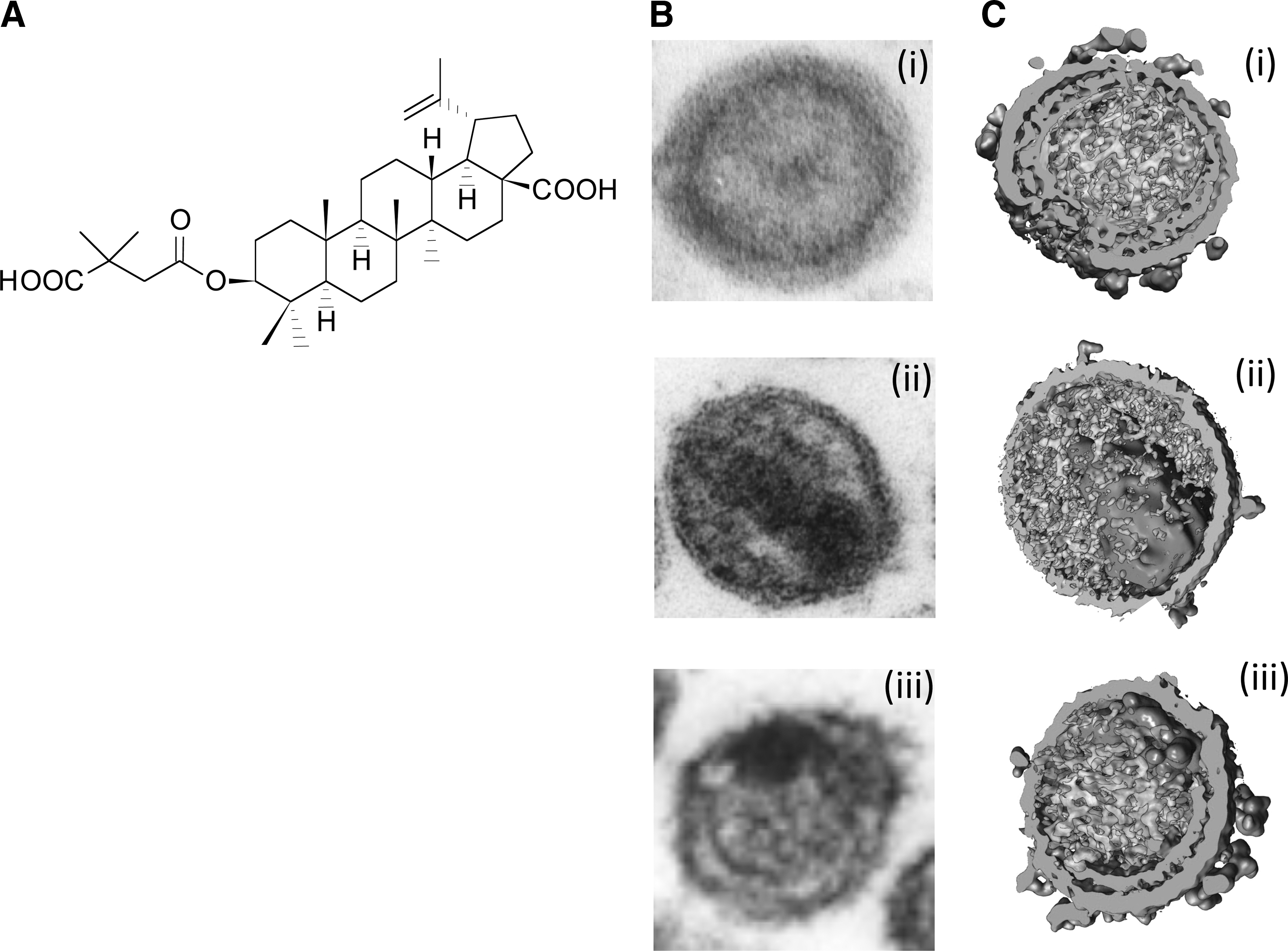
Bevirimat and its effect on HIV-1 maturation.
At the level of resolution provided by thin-section EM, the morphology of virions produced from BVM-treated cells is similar to that induced by mutations that block CA–SP1 processing. 222 By cryoelectron tomography, however, it is clear that the crescent of Gag density present in virus particles produced from BVM-treated cells (Fig. 11) displays the hexamer–hexamer spacing typical of the immature Gag lattice, suggesting that in addition to inhibiting CA–SP1 processing, BVM may stabilize the immature Gag lattice. 232
It is now clear that the CA–SP1 cleavage site in Gag is the target for BVM. Most compelling is that in vitro selection leads to the acquisition of resistance to BVM by single-amino-acid substitutions in the immediate vicinity of the CA–SP1 cleavage site (Fig. 12) rather than elsewhere in Gag or in PR. 229 –231,233 –236 In addition, mutations engineered into the CA–SP1 boundary region can confer resistance to BVM. 236 Although it is well established that BVM targets the CA–SP1 junction, the exact mechanism by which the compound inhibits CA–SP1 cleavage is not well defined.
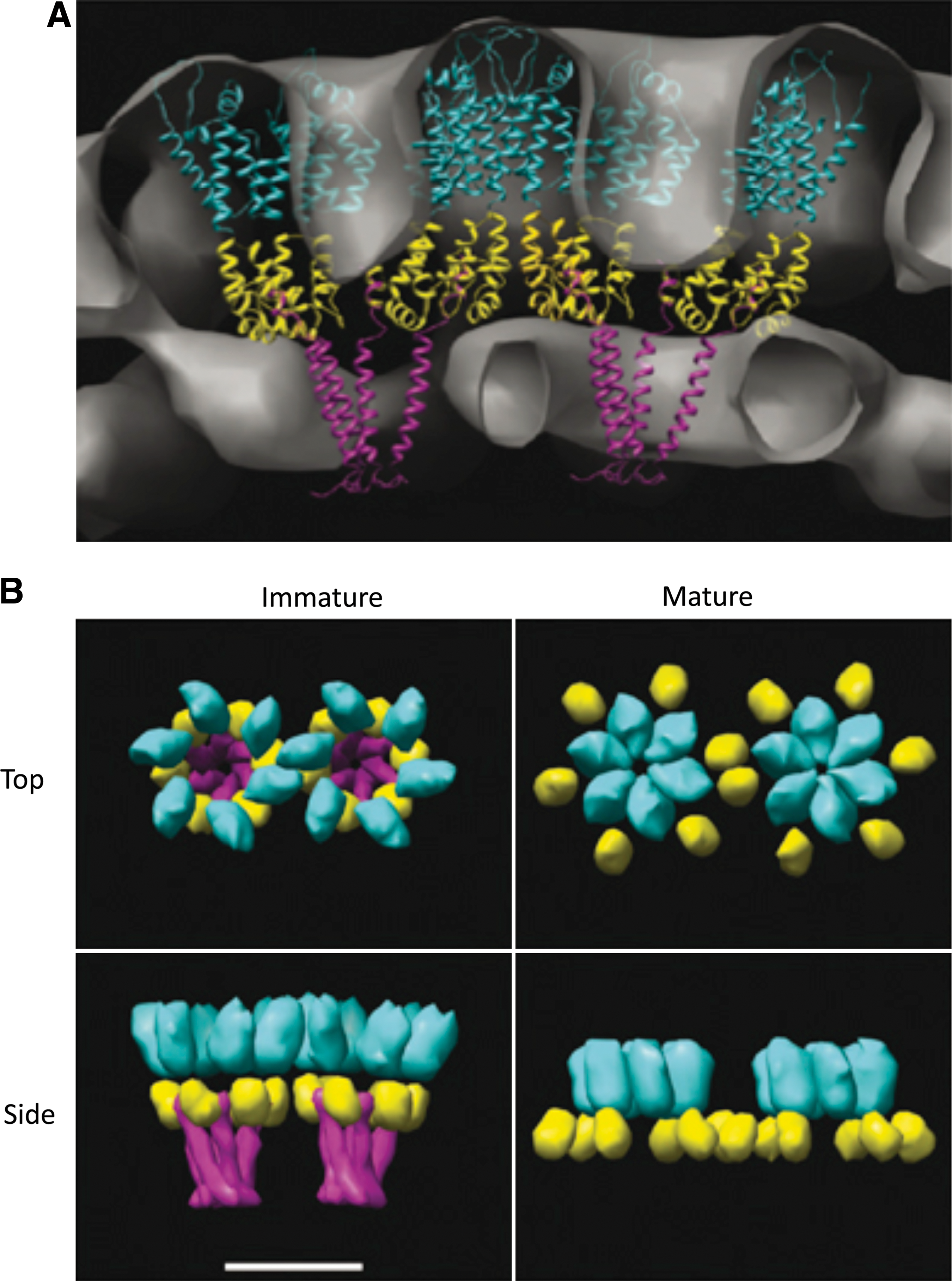
Model depicting the organization of CA in mature and immature Gag lattices.
The working model is that BVM binds a pocket on the assembled VLP that prevents access by PR to the CA–SP1 cleavage site. This hypothesis is supported by the observation that the ability of BVM to block CA–SP1 processing requires prior assembly of Gag. 230,237 Furthermore, BVM binds to immature VLPs but not mature virions, suggesting that the binding site is destroyed by PR-mediated Gag processing 238 and a number of mutations that confer BVM resistance reduce binding of labeled compound to virions. 236 It is interesting to note that some BVM-resistant mutants display enhanced replication and maturation in the presence of BVM, suggesting that in these cases a simple compound exclusion mechanism is unlikely to explain resistance. 231
BVM is the first Gag-targeted compound to reach clinical trials. Initial studies in a severe combined immunodeficiency (SCID)-hu Thy/Liv mouse model showed promising pharmacological results and led to the initiation of clinical trials in humans. 239 After Phase I clinical trials, BVM was tested in HIV-1-infected individuals. 240,241 These Phase II trials demonstrated a dose-dependent, statistically significant reduction in viral loads. 241 However, McCallister et al. showed that ∼50% of BVM-treated patients did not exhibit a reduction in viral loads despite high levels of BVM in the plasma. 242 The lack of response appeared to correlate with baseline polymorphisms in SP1 residues 6–8, 242 –244 and in vitro studies demonstrated that mutations in these residues can decrease the susceptibility of HIV-1 to BVM. 233,245 These naturally occurring Gag polymorphisms are distinct from the resistance mutations selected by passaging HIV-1 in the presence of BVM in vitro. 231,234 Although some mutations in SP1 residues 6–8 reduce sensitivity to BVM, mutations in PR that render the enzyme resistant to PIs can delay the acquisition of BVM resistance in vitro. 234
Although BVM has demonstrated proof-of-principle that CA-SP1 cleavage can be targeted to interfere with HIV-1 replication in patients, commonly occurring polymorphisms in SP1 that reduce drug susceptibility pose a challenge for future clinical development of this compound. Research performed at Pfizer has identified a small molecule that, like BVM, blocks CA–SP1 processing. Interestingly, this compound is structurally quite distinct from BVM, even though mutations at the CA–SP1 junction can confer resistance to both compounds. 246 Advances in structural biology that would allow visualization of the BVM binding site on the Gag oligomer would make possible rational, structure-based approaches to the design of more potent and broadly active inhibitors that target CA–SP1 processing.
The observation that incomplete processing at any of the Gag cleavage sites is highly detrimental to virion maturation raises the possibility that unprocessed Gag fragments can act in a dominant-negative manner to interfere with maturation and infectivity. This issue was addressed recently in several studies, which demonstrated by using low doses of PR inhibitors or a genetic approach involving cotransfection of wild-type and cleavage-site-mutant molecular clones that uncleaved Gag fragments do indeed disrupt HIV-1 infectivity. 247 –249 These results highlight the potential for drugs that block virus maturation by disrupting individual steps in the Gag processing cascade.
Uncoating and nuclear import
Thus far, our discussion has been focused primarily on targeting Gag during the late steps in the virus replication cycle. However, Gag domains also performs functions at early, postentry steps. As discussed previously, after the fusion of the viral and cellular membranes, the viral core is delivered into the cytoplasm. A poorly understood process known as uncoating then takes place; during this process, the viral RNA is reverse transcribed to double-stranded DNA. 3 Although the details of the uncoating mechanism remain to be elucidated, it is clear that correct regulation of core uncoating is required for productive infection to take place. CA mutants that display either premature or delayed uncoating in in vitro assays display impaired reverse transcription and infectivity. 250 –252
The importance of core uncoating has been underscored by the discovery of host restriction factors that target CA during early steps in the retroviral replication cycle. 9 The classical example of such a host restriction factor is Fv1, shown decades ago to restrict infection by certain strains of MLV 253 and more recently demonstrated to encode a Gag-like product derived from an endogenous retrovirus. 254 Residues in CA regulate susceptibility to Fv1, 255 implicating CA as the primary target of Fv1 restriction. A somewhat analogous restriction factor was found to function in primate cells to restrict infection by a variety of lentiviruses. In 2004, Stremlau and colleagues identified this restriction factor as TRIM5α (tripartite motif 5, isoform α). 256
Soon thereafter, a related gene in which TRIM5 is fused to cyclophilin A (hence named TRIMCyp) was identified as a restriction factor in owl monkey cells. 257,258 TRIM proteins contain a RING finger, a B-box, and a coiled-coil domain 9 ; the B-box and coiled-coil domains are required for TRIM multimerization and anti-HIV-1 restriction activity. 259,260 The α isoform of TRIM5 contains an additional C-terminal domain referred to as PRYSPRY (or B30.2); it is this region of the protein that possesses CA-binding properties. In the case of TRIMCyp, the PRYSPRY domain is replaced by cyclophilin A, which, as discussed earlier, binds the CA NTD. 256,261 –267
Although the exact mechanism by which TRIM5α and TRIMCyp exert their antiviral activity is not fully understood, CA is the target for restriction. It appears that the TRIM-family restriction factors bind the incoming viral capsid complex and promote rapid and premature uncoating and, under some conditions, proteasomal degradation. 265,268 –272 The antiviral activity of TRIM5α is species-specific, i.e., TRIM5α from rhesus macaques blocks HIV-1 infection, whereas human TRIM5α is only weakly active against HIV-1 but strongly inhibits infection by some strains of SIV. 9 Recent structural analysis indicates that TRIM5α assembles into a flexible, hexagonal lattice that uses the CA hexagonal array as a template. 273
Because the assembly of hexagonal arrays of CA appears to be a highly conserved feature of retroviral core structure, such a model helps to explain how TRIM family proteins can restrict a wide array of distantly related retroviruses.
The potent antiviral activity conferred by TRIM5α raises the possibility that these host restriction factors could in some way be harnessed to interfere with HIV-1 replication. Akkina and co-workers used a gene therapy approach to deliver an HIV-1-restricting human-rhesus TRIM5α chimera in a SCID-hu mouse model system. 274 Transgenic thymocytes and macrophages were refractory to HIV-1 infection in vivo in this setting, illustrating the potential for TRIM-based antiviral strategies. 274 Since gene therapy approaches face many hurdles in the effective treatment of HIV-1 infection in humans, alternative strategies for exploiting TRIM5α should be considered. These include the possibility that small molecules could be designed to alter human TRIM5α conformation such that it acquires the capacity to bind and restrict incoming HIV-1 core complexes. Any efforts to modify or emulate the TRIM5α-mediated block will require more information on the structural details of the TRIM–CA interaction and a more in-depth understanding of the mechanism by which this family of proteins restricts viral infection.
The import of the PIC into the nucleus represents an additional postentry step in the replication cycle that could be targeted by antiretroviral agents. This step has been difficult to study, and the viral determinants of nuclear import have proven elusive. Mounting evidence suggests that CA plays an important role in nuclear import 12,275 and a recent study developed an inhibitor of this process. KewalRamani and colleagues reported that a cytosolically localized fragment of cleavage and polyadenylation factor 6 (CPSF6) potently inhibits HIV-1 infectivity by blocking nuclear import of the PIC. 276 Selection for resistance to the CPSF6 fragment revealed that a single-amino-acid change in the CA NTD provides escape from the restriction. Remarkably, the CFSF6-resistant CA mutant displays an altered specificity in terms of its requirement for nuclear import factors, suggesting that CA may interact directly with components of the nuclear pore. 276,277 An increased understanding of this putative interaction could lead to novel approaches for blocking nuclear import of the HIV-1 PIC, a step in the replication cycle that is particularly critical in nondividing cells such as monocyte-derived macrophages.
Conclusions
In recent years, the HIV field has experienced an explosion of new information on the role of Gag proteins in HIV-1 assembly, release, and maturation. New insights have also been gained on the function of Gag proteins, in particular CA and NC, early in the replication cycle. Advances in structural biology have added greatly to our knowledge of Gag–Gag interactions and developments in cell biology have revealed the molecular details of host cell pathways that the virus uses to promote its replication and cellular restriction factors that impede virus infection. Despite these impressive advances, Gag proteins have not yet been successfully exploited in the development of clinically approved antiretroviral therapies. Although current anti-HIV drugs are highly effective in most patients, over time it is likely that resistance to these inhibitors will become an increasing problem. We therefore believe that attention should begin to broaden from basic studies on Gag function to the application of this information to novel inhibitor development.
Footnotes
Acknowledgments
We thank L. Henderson for providing the image shown in Fig. 1 and M. Summers for ![]() . We also thank W. Sundquist, M. Summers, E. Wright, A. Steven, M. Yeager, J. Hurley, and G. Jensen for permission to reproduce figures and members of Freed laboratory for helpful discussions and critical reading of the manuscript. Research in Freed laboratory is supported by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research, and the Intramural AIDS Targeted Antiviral Program.
. We also thank W. Sundquist, M. Summers, E. Wright, A. Steven, M. Yeager, J. Hurley, and G. Jensen for permission to reproduce figures and members of Freed laboratory for helpful discussions and critical reading of the manuscript. Research in Freed laboratory is supported by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research, and the Intramural AIDS Targeted Antiviral Program.
Author Disclosure Statement
No competing financial interests exist.