
Abstract
Several nonhuman primate models are used in HIV/AIDS research. In contrast to natural host models, infection of macaques with virulent simian immunodeficiency virus (SIV) isolates results in a disease (simian AIDS) that closely resembles HIV infection and AIDS. Although there is no perfect animal model, and each of the available models has its limitations, a carefully designed study allows experimental approaches that are not feasible in humans, but that can provide better insights in disease pathogenesis and proof-of-concept of novel intervention strategies. In the early years of the HIV pandemic, nonhuman primate models played a minor role in the development of antiviral strategies. Since then, a better understanding of the disease and the development of better compounds and assays to monitor antiviral effects have increased the usefulness and relevance of these animal models in the preclinical development of HIV vaccines, microbicides, and antiretroviral drugs. Several strategies that were first discovered to have efficacy in nonhuman primate models are now increasingly used in humans. Recent trends include the use of nonhuman primate models to explore strategies that could reduce viral reservoirs and, ultimately, attempt to cure infection. Ongoing comparison of results obtained in nonhuman primate models with those observed in human studies will lead to further validation and improvement of these animal models so they can continue to advance our scientific knowledge and guide clinical trials.
Introduction
T
Since the discovery of HIV in 1983 as the causative agent of the acquired immune deficiency syndrome (AIDS), a vast database of information has been collected on the many aspects of HIV biology, including genetic structure, transmission, and the virus–host cell interactions that play a role in the pathogenesis. 2,3 Although behavioral interventions (including education and provision of condoms) significantly reduce transmission, they are insufficient to halt the epidemic. Accordingly, there has been an enormous global effort to develop biomedical antiviral strategies to prevent and/or treat infection, of which the success rates have been highly variable.
Although much effort has focused on developing an HIV vaccine, none of the candidates that have been tested in human trials so far was found to be highly effective. 4 There is, however, great enthusiasm—as evidenced by the recently completed and the many ongoing trials—to give uninfected people who are at high risk of acquiring infection access to antiretroviral drugs that reduce their likelihood of infection. This includes the oral administration of antiretroviral drugs starting before or shortly after exposure (so-called preexposure and postexposure prophylaxis) to achieve systemic drug levels, or the topical application of antimicrobial products, so-called microbicides, onto the vaginal or rectal mucosa. 5,6
In contrast to the limited success of vaccines, major progress has been achieved in the clinical management of HIV-infected people due to the growing arsenal of antiretroviral drugs that target various steps in the viral replication cycle, and that are given as combinations in highly active antiretroviral therapy (HAART; ART). For many HIV-infected people, ART has turned HIV infection into a chronic manageable disease. 7,8 In addition, it was also found that treating HIV-infected persons on ART has a very dramatic impact on reducing the transmission rates to their partners. 9
Despite the considerable success of ART, there is still room for improvement. Long-term administration of many drugs is often complicated because of issues such as cost, storage, toxicity, compliance, and drug resistance; some of these problems are more pronounced in developing countries. Accordingly, novel compounds continue to be developed. 10,11 In addition, the quest continues to find strategies to boost immunological control to reduce the need for antiretroviral drugs, or even more ambitiously, to purge out viral reservoirs and ultimately cure HIV infection, so that antiretroviral drug treatment can be withdrawn permanently. 12
The pipeline that antiviral products need to cross before the intended approval for clinical use is tedious, time-consuming, and very expensive. The majority of antiretroviral drugs, microbicides, and vaccines that look initially promising fail to reach clinical approval because a lack of resources prevents their development, or because during the sequential process of preclinical and clinical testing, they show unfavorable immunogenicity, pharmacokinetics, and toxicity, or insufficient efficacy. 13 In addition, a clinical trial aimed at reducing HIV infection rates generally needs to enroll and follow very large cohorts of high-risk participants for several years to determine efficacy, which due to the limited resources often means that only a limited number of trials can be performed simultaneously. In contrast, trials that test the therapeutic effects of a compound on established HIV infection can usually determine in a relatively short time with smaller cohorts whether further testing is warranted.
The correlation between in vitro effects and in vivo safety and efficacy to treat or prevent infection is often weak. The desired outcome of a product in vivo—protection against infection or against disease—is determined by many complex interactions between the virus, the host, and the compounds, most of which cannot be mimicked appropriately by in vitro studies or mathematical models. This is where animal models can be beneficial (Fig. 1). The development of antiviral products can be accelerated by efficient and predictive animal models capable of screening and selecting the best compounds at each decision point (Fig. 2). Although regulatory agencies request safety/toxicity data from animal models, they do not require efficacy data from animal models before allowing a novel compound to enter clinical trials. Accordingly, an opportunity to discard ineffective drugs early in the development process (and thus save much time and resources) is lost whenever an available appropriate animal model of efficacy is bypassed. 14 In addition to showing proof-of-concept of efficacy, such animal models can also be extremely useful to define surrogate markers of efficacy (e.g., immune correlates of protection by vaccines), which is valuable not only to better understand the mechanisms of action, but also to streamline and accelerate future product development.
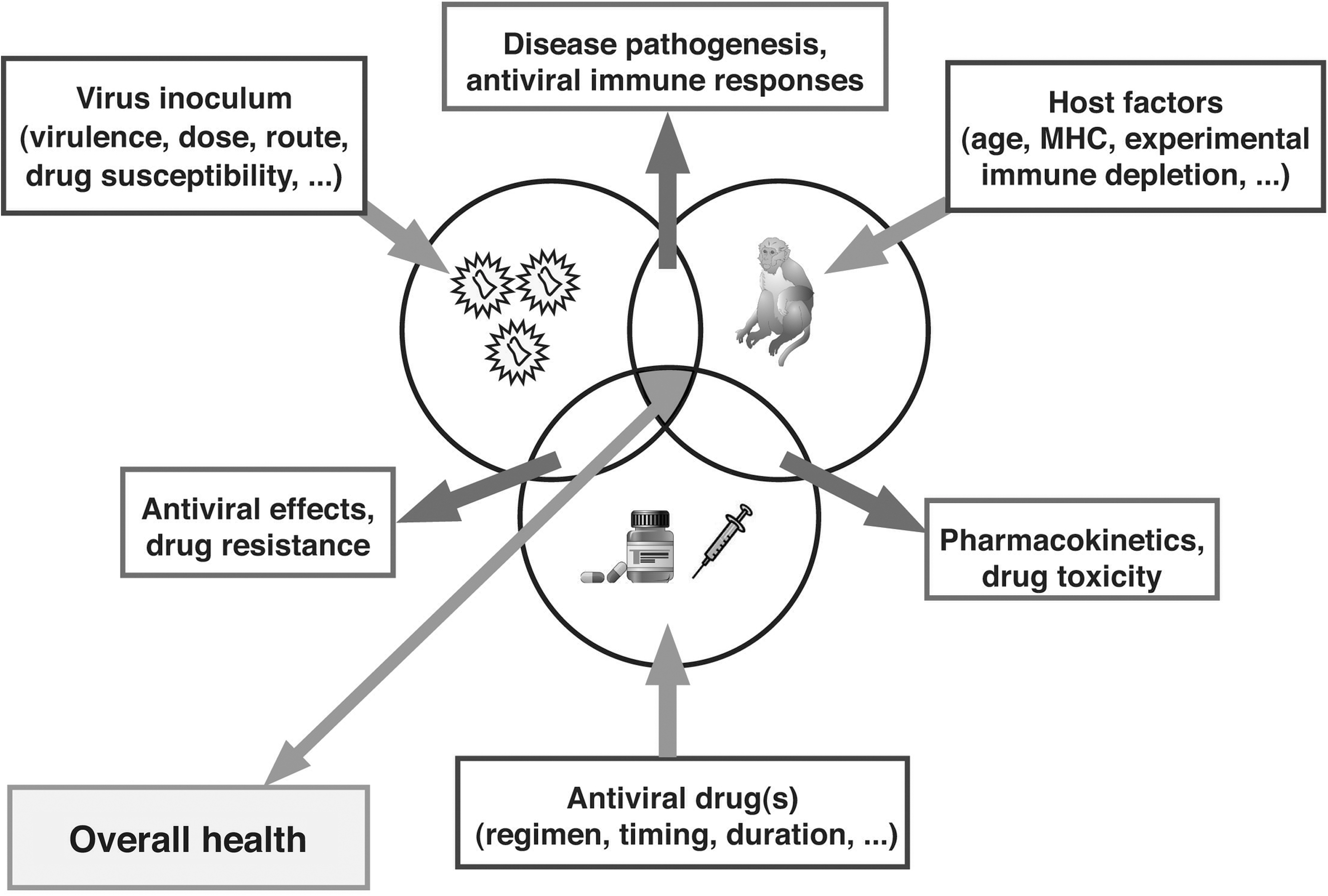
Determinants of the outcome of antiviral drug treatment. The ultimate goal of antiviral strategies is to improve or maintain the overall health of the host by preventing infection or disease progression. This outcome is determined by many variables and interactions between the virus, the host, and the antiviral drugs, most of which cannot be mimicked appropriately by in vitro studies. Animal models can be very valuable, not only for preclinical screening of compounds (see Fig. 2), but also to acquire better insights into pathogenesis and mechanisms of protection. Animal models allow us to control and manipulate many input variables through experimental approaches that are not feasible or ethical in humans (such as in vivo depletion of certain immune cells, inoculation of animals with drug-resistant mutants, or drug monotherapy). Modified from Van Rompay. 20
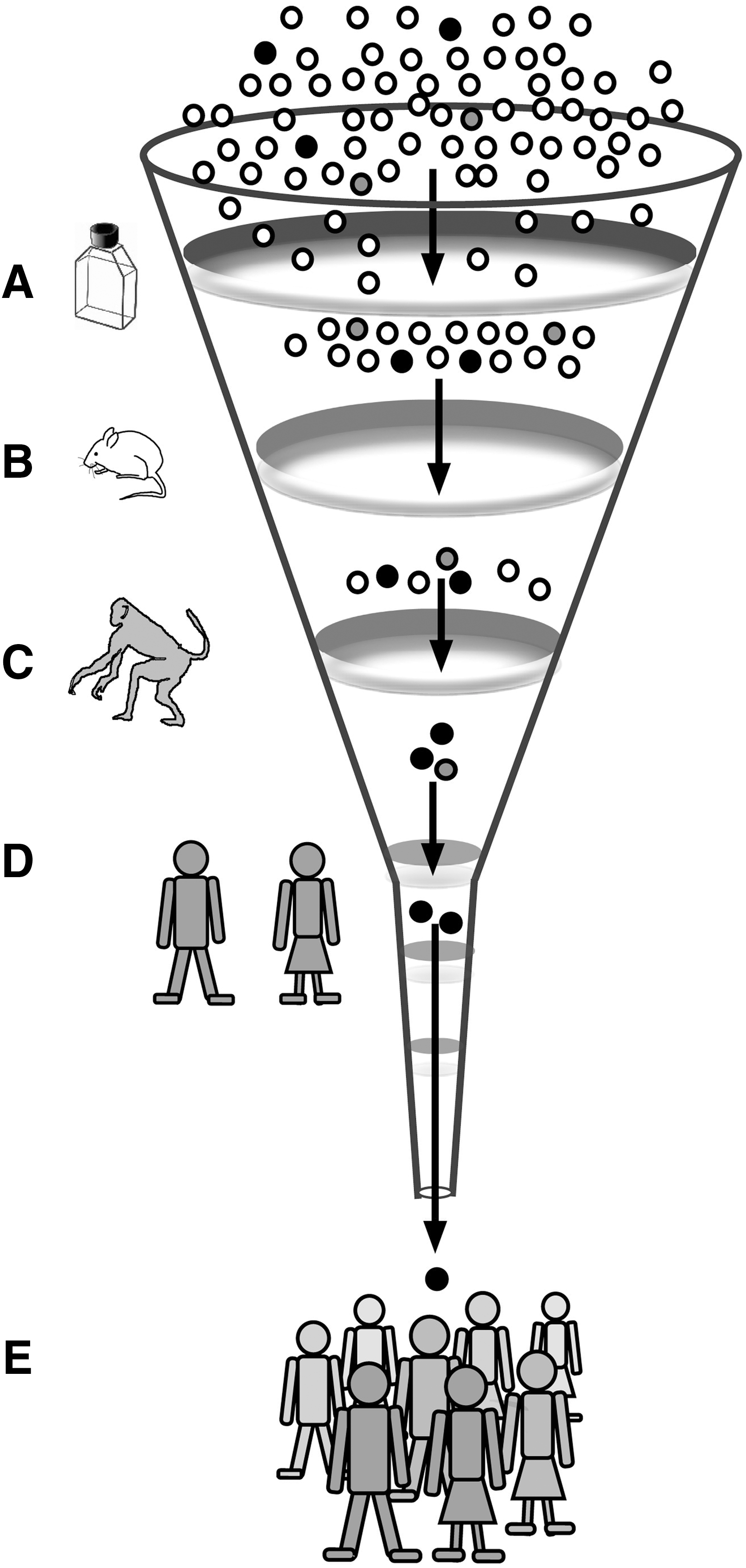
Role of animal models in the development of anti-HIV strategies. Because human trials are very expensive, time-consuming, and logistically very complicated, only a limited number of antiviral strategies can be evaluated. Progress can be accelerated by prescreening efficacy and toxicity in several steps. High-throughput systems with relatively low cost, such as in vitro testing
In addition to being a test system to screen novel antiviral regimens, animal models can also be used to explore novel hypotheses that are logistically or ethically difficult to explore in humans. By manipulating the input variables and experimental design (e.g., the age of the animals; the virulence and drug susceptibility of the virus inoculum; the composition, timing, and duration of the drug regimen), studies can be performed to unravel specific questions on the many virus–host–drug interactions. 15 As discussed further in this review, examples of such studies are those focused on evaluating the in vivo virulence and clinical implications of drug-resistant viral mutants, and the role of antiviral immune responses on antiviral drug efficacy.
The ideal animal model of HIV infection would be one that involves HIV infection of a small animal and that closely mimics HIV transmission, pathogenesis, and the effects of antiviral strategies. However, because such an animal model does not yet exist, it is important to acknowledge that each of the currently available animal models has its limitations.
Although several lentiviruses (maedi-visa virus, caprine arthritis encephalitis virus, and equine infectious anemia) cause slowly progressive degenerative diseases of certain farm animals, a major difference with HIV infection of humans is that these lentiviruses infect only the macrophages and monocytes and not CD4+ T helper cells and, therefore, they do not cause immunosuppression in their hosts; this limits their relevance as a model for HIV immunopathogenesis. 16 –19 A variety of rodent and feline models of HIV infection have been developed; some of the rodent models have been engineered to be permissive to HIV-1 replication. 20 While these rodent and feline models are useful for initial screening (Fig. 2), each has its own limitations because of significant differences in host physiology (including immunology and metabolism), viruses, pathogenesis, or disease outcome. 20 Further testing is done best in nonhuman primate models that more closely resemble HIV infection of humans and therefore allow a more reliable extrapolation of results of antiviral strategies. Accordingly, nonhuman primate models of HIV are the focus of the remainder of this review.
Overview of Nonhuman Primate Models for HIV Disease and Control
Nonhuman primates are phylogenetically the closest to humans, and have very similar physiology, including immunology and pharmacology. A common theme among the different nonhuman primate models is that disease, which resembles AIDS in humans, is most consistently seen during infection of nonnatural hosts.
Chimpanzees in the wild are the source of SIVcpz, the immediate precursor of HIV-1. Although SIVcpz infection of chimpanzees in the wild is associated with an increased mortality rate, 21 very few animals that have been experimentally infected with SIVcpz or HIV-1 in captivity have developed disease. 22 –24 In addition, this animal model is not practical due to the low availability, its high price, and ethical issues. 22 HIV-1 infection could be induced in young pigtailed macaques, but virus replication was not sustained and no disease was observed. 25 HIV-2 infection models have been developed with hamadryas baboons (Papio hamadryas) and several macaque species; depending on the HIV-2 isolates, the outcome varied from an AIDS-like disease with CD4+ T cell decline to no disease. 26 –28
Many nonhuman primate species in Africa are naturally infected with simian immunodeficiency virus (SIV) strains; examples are African green monkeys (SIVagm) and sooty mangabeys (SIVsm). These viruses are more closely related to HIV-2 than to HIV-1. Despite persistent high-level virus replication, these natural hosts rarely develop disease. Accordingly, these natural, nonprogressive SIV infections represent an evolutionary adaptation that allows a peaceful coexistence of primate lentiviruses and the host immune system; research indicates that this adaptation involves phenotypic changes to CD4+ T cell subsets, limited immune activation, and preserved mucosal immunity, all of which contribute to the avoidance of disease progression. 29 –33 In contrast, due to cohousing different primate species, 34 it was discovered coincidentally in the mid-1980s that SIV infection of nonnatural hosts such as Asian macaques results in a disease that resembles human AIDS in many aspects. 35 While limitations of the SIV-macaque models remain their high cost, relative availability, and the subtle differences between HIV and SIV (i.e., SIV resembles genetically and structurally HIV-2 more than HIV-1), the many similarities in virus, host, and disease pathogenesis have made them the currently premier animal model in HIV research.
Main Features and Development of the SIV-Macaque Models
The outcome of infection depends on a complex interaction of host and viral determinants. The most commonly used species are rhesus macaques (Macaca mulatta), cynomolgus macaques (M. Fascicularis), and pigtailed macaques (M. nemestrina). Additional host factors within a species that have been observed to affect transmission and disease pathogenesis include MHC I alleles (e.g., Mamu-A*01, Mamu-B*08) and restriction alleles (such as TRIM5α). 36 –39
The SIV isolates generally belong to a few groups, in particular SIVmac, SIVsm, and SIVmne. While CD4 is their main cell receptor, most use CCR5 as a coreceptor. Infection of macaques with virulent SIV isolates such as uncloned SIVmac251 and molecularly cloned SIVmac239 results, after an asymptomatic period, in a disease that resembles human AIDS in many aspects, including cell tropism, generalized immune activation, CD4+ T cell depletion (especially from mucosal sites), opportunistic infections, weight loss, and wasting. 35,40 However, it is important to remember that SIV infection of macaques is not necessarily fatal, because based on the selection of the host and the SIV isolate or clone, a broad spectrum of clinical outcomes is observed. At one end of the spectrum are models in which highly virulent SIV isolates induce persistently high viremia and rapid disease progression within a few months (e.g., SIVmac251 infection of newborn macaques); at the other end are avirulent isolates that induce transient or low-level viremia and no disease, even in newborn macaques. 41 This spectrum of infection outcomes makes these animal models also suitable to assess how genetic changes in the virus (e.g., mutations that confer drug resistance or immune escape) affect viral virulence.
To mimic the common routes of HIV transmission in humans, nonhuman primate models have been developed for different routes of virus inoculation (i.e., intravenous, vaginal, penile, intrarectal, or oral). 42 –45 In addition, pediatric AIDS models have been developed by using pregnant and infant macaques. 41,46 –48
To achieve high infection rates, two main models are used for the different inoculation routes; while the earlier studies generally used a single high-dose inoculation, more recently models with repeated low-dose inoculations have been developed. 49 The repeated low-dose mucosal inoculation models recapitulate many of the features of mucosal HIV transmission—such as transmission of a limited number of variants across the mucosal barrier and early diversification—better than the high-dose models. 50 –52 The repeated low-dose inoculation models are particularly relevant to test prophylactic strategies in which efficacy may be missed with the high-dose inoculation models. 52,53
In the early 1990s, the SIV macaque model was already successful in demonstrating some vaccine efficacy, particularly with live-attenuated SIV vaccines. 54,55 In contrast, during those early years, the practicality of the SIV-macaque model to test antiviral compounds was limited, and most drug studies were not successful. Some critics interpreted this as an indication that the animal model was not appropriate for antiviral drug studies. However, viewed retroactively, a combination of factors was responsible for these observations. First, the drugs that were available then were relatively weak, had very complicated dosage regimens, or were toxic, which made long-term administration difficult. In addition, there were few means to monitor infection and disease progression. The measurement of p27 antigen or infectious virus in plasma or peripheral blood mononuclear cells (PBMCs) was not sensitive enough to detect any minor changes in viral replication, particularly during the asymptomatic stage of SIV infection. Because the rate of CD4+ T cell decline and disease progression can be quite variable, the relatively small animal numbers that were used made it often difficult to determine whether a small difference in clinical outcome was due to host factors or to the intervention when the drug had only poor or modest antiviral activity. Accordingly, the development of quantitative and highly sensitive assays to measure viral RNA levels in plasma in infected macaques, the observations that plasma RNA levels were predictive of disease progression, and the demonstration that more potent antiviral drugs were able to reduce virus levels and delay the disease course were big milestones. 56 –59 Now the effects of antiviral strategies on SIV infection and disease progression can be monitored by the same laboratory markers (particularly viral RNA levels in plasma and CD4+ T lymphocyte counts) as those that are used to monitor HIV-infected people (Table 1).
PCR, polymerase chain reaction; RT-PCR, reverse transcriptase-polymerase chain reaction; ELISA, enzyme-linked immunosorbent assay; ADCC, antibody-dependent cell-mediated cytotoxicity; ADCVI, antibody-dependent cell-mediated virus inhibition; NK, natural killer.
Although SIV is related to HIV-1, there are still many genetic differences that need consideration. Preclinical testing of antiviral strategies in the SIV-macaque model is most relevant if the viral target and its susceptibility to inhibition naturally resemble, or can be engineered to resemble that of HIV-1. Because most SIV isolates use the CCR5 chemokine receptor, they are also susceptible to some CCR5-targeting entry inhibitors. 60 Because of much similarity in the polymerase region, SIV is also susceptible to many nucleoside and nucleotide reverse transcriptase (RT) inhibitors, integrase inhibitors, and protease inhibitors. 61,62 In contrast, most nonnucleoside RT inhibitors (NNRTIs) such as nevirapine and efavirenz are active only against HIV-1 and not against HIV-2 or SIV. 61 Accordingly, the construction of infectious SIV/HIV-1 chimeric viruses, in which the RT gene of SIV was replaced by its counterpart of HIV-1 (so called RT-SHIVs) has allowed evaluation of NNRTIs in primate models 63 (Fig. 3).
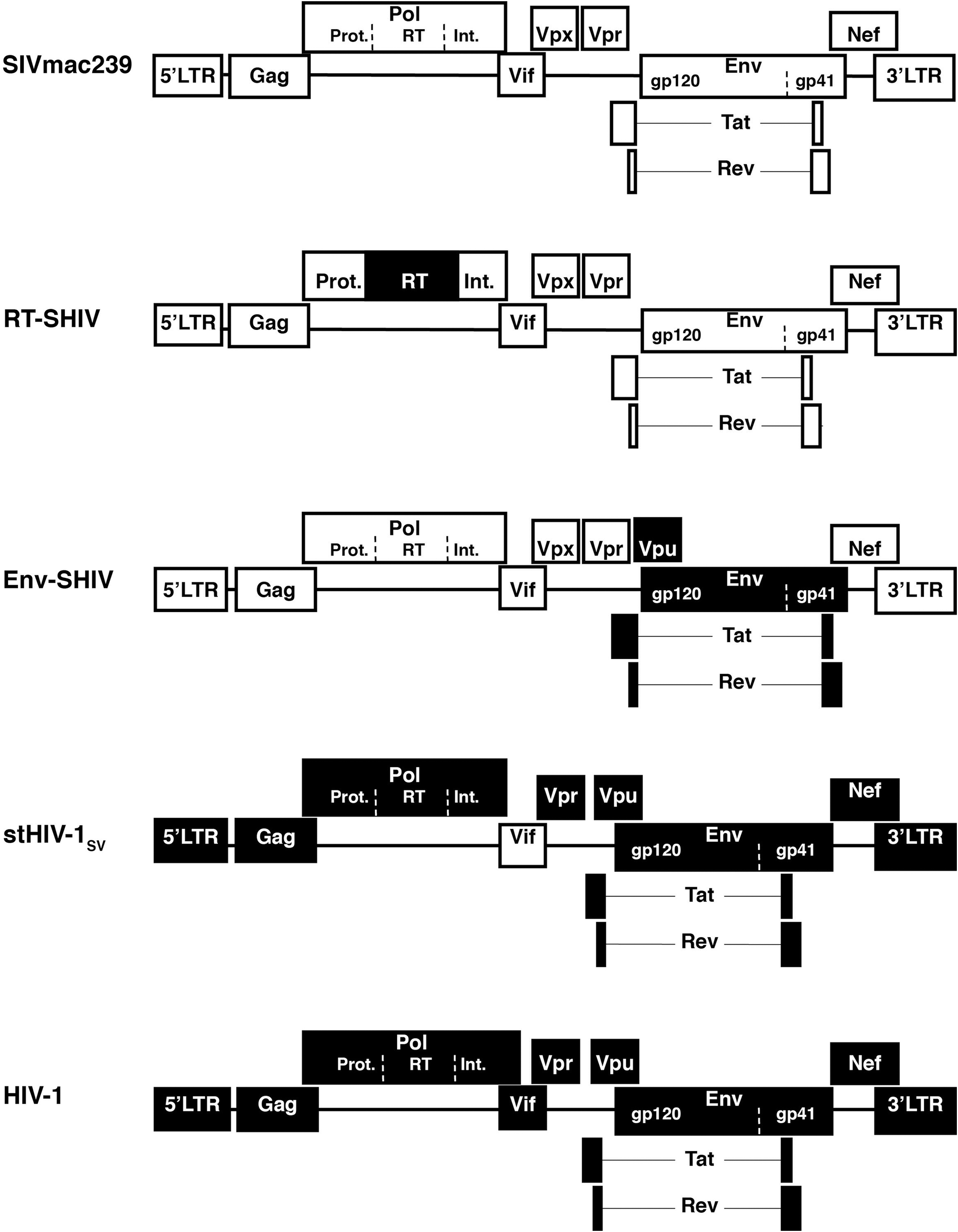
Schematic representation of SIV, HIV-1, and chimeric constructs that are used in nonhuman primate studies. Because there are important genetic differences between SIV and HIV-1 that include the targets of some antiviral strategies, chimeric viruses have been constructed that, depending on the construct and further in vivo passaging, can induce transient to persistent viremia and sometimes disease. In this schematic representation, white and black boxes refer to SIV and HIV sequences, respectively. Reverse transcriptase simian-human immunodeficiency virus (RT-SHIVs) contain HIV-1 reverse transcriptase (RT) in an SIVmac239 background 63 or SIVmne background. 279 Env-SHIVs consist of SIV background containing HIV-1 env, tat, rev, and generally (to have better replication in macaques) also vpu. 64 Simian-tropic (st) HIV-1 strains differ from HIV-1 only in the vif gene. 74
Similarly, because there are significant differences between SIV and HIV envelope, env-SHIVs that contain the HIV-1 envelope region have been constructed to allow direct testing of strategies that target the HIV-1 envelope region (Fig. 3). Many env-SHIVs are attenuated but some virulent isolates have been derived through serial passage. Most virulent env-SHIVs such as SHIV-89.6P, while useful to address specific questions, have the limitation that their disease pathogenesis (including CXCR4 coreceptor usage and very rapid CD4+ cell depletion) differs from the typical course seen with HIV and SIV infection. 64 –68 Most of the currently available CCR5-using env-SHIVs (such as SHIV-SF162P, SHIV-SF162P3, and the clade C env-SHIV-1157i) have the limitation that after the initial peak of viremia, a significant portion of untreated animals suppressed viremia to low or undetectable levels and did not develop disease 69 –71 ; although such viruses can be useful to test prophylactic or early postinfection interventions, this large variability in chronic viremia and disease outcome makes them less suitable to test the efficacy of antiviral strategies during established infection, especially when animal numbers are limited. Attempts to increase the virulence of these env-SHIV models are already fruitful, as the further passaged virus SHIV-SF162P3N gives persistent viremia and disease more consistently. 72,73
In ongoing attempts to use a virus that resembles HIV-1 as much as possible, investigators constructed recently simian-tropic (st)HIV-1 strains that differ from HIV-1 only in the vif gene (Fig. 3); these viruses caused persistent viremia in pigtailed macaques for several months after which viremia was controlled by the immune system. 74 Ongoing development of these stHIV-1 models is likely to lead to a more persistent viremia.
Contributions of the Nonhuman Primate Models to HIV Vaccine Development
Since its early development, the SIV animal model has been used extensively to test different vaccine approaches and to attempt to define their correlates of protection. The very limited success of HIV vaccine candidates in phase III trials in humans precludes proper validation of the vaccine data obtained in the different nonhuman primate models. While many excellent review articles on the use of primate models for preclinical HIV vaccine development have been published, 75 –78 this review will focus on highlighting some general themes and lessons that reflect the current status, with the caveat that this is subject to continuing changes in coming years. 79
Progress toward developing an HIV vaccine has been hampered by the uncertainty about what host immune responses are required for efficacy. An ideal HIV vaccine would provide sterilizing immunity (i.e., completely prevent infection). However, a more realistic goal that is consistent with the mode of action of other viral vaccines is that while HIV vaccine-induced immune responses may not prevent the initial infection, they may prevent or reduce systemic dissemination, limit viral replication permanently, and protect against disease progression.
A large variety of vaccines have been tested in nonhuman primate models. In general, live-attenuated vaccines have been most successful, probably because they generate persistent and broad antiviral immune responses, 54,55,80 but due to safety concerns this approach is currently not being explored in humans. Many other vaccine approaches—including DNA, replicating and nonreplicating vectors, subunit proteins—have been tested, including in a variety of prime-boost regimens. Although many of these regimens have shown various levels of efficacy in macaque models, no single in vitro immune effector function or combination of markers has been consistently associated with efficacy. In other words, different vaccine strategies may achieve some level of efficacy through different mechanisms, and the outcome also varies often depending on host genetics (such as MHC alleles or restrictive TRIM5 alleles) and the selection of the viral challenge model (including strain, dose, and route of inoculation). 80 –82
Many passive immunization studies have demonstrated that antibodies with high antiviral activity in vitro can confer protection (“sterilizing immunity”) against mucosal infection in infant or adult macaques. 83 –88 Although the antibodies that were used had generally high neutralizing activity against the challenge virus in vitro, protection could also be observed with antibodies that had no neutralizing activity but had high antiviral activity in an antibody-dependent cell-mediated virus inhibition (ADCVI) assay. 87,89 Although these passive immunization studies provide important proof-of-concept of the prophylactic potential of antibodies, it has been very difficult to induce antibodies with broadly neutralizing activity through active immunization. 90 This is exemplified by the failure of envelope-based subunit vaccines to protect macaques against heterologous virulent challenge viruses 77 ; these results are consistent with the failure of the Vaxgen's gp120-based AIDSVAX HIV-1 vaccines in humans. 91
Studies in macaques that investigated the role of antibodies during primary or chronic viremia have had various results—ranging from beneficial to adverse effects—depending on the experimental conditions. 15,87,92 –95 In contrast, numerous studies, including some that used in vivo CD8+ cell depletion, have demonstrated that CD8+ T cell responses play an important role in determining peak viremia and viral set point after infection. 81,96,97 Thus, immunologic mechanisms that mediate protection against initial infection may be different from those that control virus replication after infection. These results provided the impetus for the development of vaccine strategies that induce both arms—robust antibody and CD8+ cell-mediated immune responses.
However, in the past few years, the quest of the HIV vaccine research field to develop an effective vaccine and identify immune correlates of protection has been stirred up significantly by the results of two phase III clinical trials, the STEP Study (which failed to show protection) and the Thai ALVAC/AIDSVAX trial (which found a 30% reduction in infection rate). 4,98 The comparison of the outcome of these vaccine trials with data obtained in nonhuman primate models has provided very valuable clues that need to guide future research.
In the STEP human trial, an adenovirus type 5 (Ad5)-based HIV vaccine designed to induce cell-mediated immune responses was quite effective in inducing such immune responses but failed to either prevent HIV-1 infection or suppress viremia in subsequently infected participants, and may even have enhanced infection rates in subgroups. 98 Despite these disappointing results, the outcome of the STEP Study provided some very important lessons, including concerning the relative predictability of the different nonhuman primate models, particularly the selection of the challenge virus. When a similar vaccine had previously been tested in macaques against CXCR4-tropic SHIV89.6P, it did not protect macaques from infection, but caused a substantial reduction in viral load and a preservation of CD4+T cell counts after infection. 99 In contrast, against virulent CCR5-tropic SIV, an analogous vaccine failed to either protect against infection or reduce the viral set point. 100 Based on these results, CXCR4-tropic pathogenic SHIVs such as SHIV89.6P are now considered inadequate predictors of efficacy in humans; the virulent SIV challenge model, although still incompletely validated, is currently considered to be the more relevant one to filter vaccine candidates through the pipeline, because it recapitulates more closely (albeit still imperfectly) the key features of HIV infection and pathogenesis. 68,78
In the recently completed RV144 trial in Thailand, a vaccine regimen consisting of recombinant canarypox vector expressing HIV (ALVAC-HIV) and recombinant gp120 (AIDSVAX) demonstrated despite relatively low/moderate immunogenicity (as defined by the currently used immune assays) a moderate protection against infection particularly in low-risk groups, but did not affect virus levels in individuals who became infected. 4 Similarly, ALVAC-SIV had low immunogenicity, but when tested in an infant macaque model with repeated low-dose SIV exposures gave partial protection against infection and had only a small, albeit insignificant effect on reducing viremia for those animals that became infected. 53 However, ALVAC-SIV did not prevent infection from a more intense challenge exposure, although it did reduce the viral load and delay disease progression. 101,102
Altogether, although the outcomes of all these vaccine studies confirm our relatively poor understanding of antiviral immunity and the influence of host genetics, they support some new trends. 79 One of them is that “the more, the better” may not apply to the immune responses needed for an effective HIV vaccine. The available data suggest the need for an intricate balance between quantity, quality, timing, and location of the different antiviral immune responses—both adaptive and innate—as too much immune activation and inflammation can be harmful by promoting virus replication and dissemination. 103 Thus there is a need to focus future research efforts on (1) the further development and optimization of in vitro assays and markers to better capture the variety of beneficial and harmful immunological responses that occur in vivo, and (2) the more effective use of nonhuman primate models in HIV vaccine development. 2,78,79
Contributions of the Nonhuman Primate Models to Microbicide Development
In the absence of an effective HIV vaccine, there has been considerable interest in the development of microbicides, i.e., compounds that after topical application onto mucosal surfaces can exert sufficient local antiviral activity to prevent infection. The characteristics of the ideal microbicide—effective, acceptable, safe, inexpensive, available in both spermicidal and nonspermicidal formulations, long-lasting, and usable without partner knowledge—could empower and protect many women who currently lack control over their partner's sexual behavior.
Microbicide candidates include compounds that disrupt the viral membrane, maintain an acidic vaginal pH, block cellular receptors, or antiretroviral drugs that inhibit a specific viral target.
While more than 60 products have undergone preclinical evaluation as potential microbicide candidates, only a handful have so far advanced into phase III human studies. 104 With the exception of tenofovir gel, most microbicides that have been tested so far have fared very poorly, and sometimes increased susceptibility. 5,105 –108 Progress has been hampered by our limited understanding of the biological processes during mucosal HIV transmission, and the lack of established correlates of safety or efficacy that can be used in preclinical studies (including animal models) and early clinical studies to screen candidates. 107,109
Because in vitro and rodent models have major limitations, nonhuman primates remain the most relevant animal species to prescreen microbicide candidates for safety and efficacy. 110 Studies in nonhuman primates have demonstrated that topical high-dose administration of a relatively large number of compounds protected adult macaques against intravaginal or intrarectal SIV or SHIV infection at varying rates of efficacy. 111 –122
The most likely reason of this discrepancy between the relative success of microbicides in macaques and the failures of most microbicides in humans is that most macaque models used a single or short-term administration of a microbicide followed by a single atraumatic inoculation of SIV. In contrast, in human clinical trials, the repeated administration of the first-generation microbicides, which were primarily surfactants and polyanionic compounds, led to a disruption of the epithelial barrier or induced inflammation of the vaginal mucosa; this led to a lack of efficacy or worse, increased susceptibility to infection. 105 This mucosal irritation was afterward also confirmed to occur in animals. 106,110 In other words, it has become clear in recent years that prior selection criteria were inadequate for predicting efficacy and safety in vivo, and that more stringent tests and a more rational approach are now required to advance the microbicide candidates into future trials. 14 The development of better methodologies, including improvement of nonhuman primate models, is needed to study the effect of chronic microbicide exposure on mucosal health and to identify safer microbicides. 14,107,123
Because many compounds are likely to affect mucosal physiology after prolonged use, microbicide research has gradually shifted its focus to so-called second-generation microbicides, which include virus-specific inhibitors (e.g., reverse transcriptase inhibitors such as tenofovir and emtricitabine). Particular attention was given to those antiretroviral drugs that were proven to be effective in animal models when systemic drug levels were present (i.e., preexposure and postexposure prophylaxis; see further). Depending on the viral target, appropriate SIV or RT-SHIV strains were used to demonstrate the efficacy of several antiretroviral drugs as microbicides in nonhuman primates. 122,124 –126 These results were predictive of the recent results of the CAPRISA 004 trial that demonstrated partial efficacy of a 1% tenofovir gel in protecting women against HIV infection. 5
Although there have been concerns that the topical use of antiretrovirals as microbicides by HIV-infected women may induce drug-resistant virus, so far this has not been observed to be a problem in early studies with tenofovir gel. 5,127 Additional animal studies can further assess the role of drug resistance, and also explore combinations of several compounds as microbicides, because a combination is likely to be more successful than a single agent to prevent infection and overcome the problem of drug resistance.
Contributions of the Nonhuman Primate Models to Antiretroviral Drug Development
As mentioned earlier, the nonhuman primate models had to overcome some initial hurdles to make them more practical for preclinical drug development. Fortunately, many of these problems have been solved, and in the mid-1990s, in particular the studies with tenofovir provided proof-of-concept of the validity of the macaque model. Tenofovir (previously called PMPA; 9-[2-(phosphonomethoxy)propyl]adenine) was a drug that was first proven to be effective in macaques, which sparked interest to initiate clinical testing. 59,128 –132 Because of its high efficacy in HIV-infected humans, tenofovir has now become one of the most widely used drugs in ART regimens. 133
Studies in macaque models have contributed significantly to our knowledge on the many aspects of antiretroviral drug administration.
Pharmacokinetics and toxicity
Because of their similar physiology and metabolism, nonhuman primates have been useful to study the toxicity and pharmacokinetics of antiretroviral drugs, including the effects of pregnancy and drug transfer across the placenta and into breast milk. 134 –141 To better correlate pharmacokinetics with efficacy, recent studies have also used newly developed methodologies to measure intracellular levels of the active phosphorylated forms of nucleoside and nucleotide analogs. 142,143
While most studies used short-term drug administration, studies with tenofovir have demonstrated its safety during prolonged daily treatment (>1–14 years), starting at birth and continuing throughout adulthood, including pregnancy. 132,144 These safety data of tenofovir in macaques are consistent with the favorable safety profile of tenofovir observed in humans and in the Antiretroviral Pregnancy Registry, as tenofovir is currently classified as a pregnancy category B drug (no evidence of risk to humans). 145 –147 In contrast, severe birth defects were observed in an estimated 25% of infant cynomolgus macaques born to mothers taking efavirenz early in gestation at a dose that gave similar exposure as the dosage regimen in humans 148 ; this observation led to a category C pregnancy warning (Risk cannot be ruled out) from the Food and Drug Administration (FDA), but was subsequently changed to category D (Positive evidence of fetal risk) because of reported birth defects in some infants exposed to efavirenz during the first months of pregnancy. 147
Prophylaxis: prevention of infection
Many studies in nonhuman primates investigated whether oral or parenteral administration of antiretroviral drugs near the time of virus inoculation could prevent infection. Early studies were not very effective in preventing infection, but a likely reason for this was the combination of a high-dose virus inoculum, the direct intravenous route of virus inoculation, and the relative weak potency of drugs at that time. 149 –151 The proof-of-concept that antiretroviral drugs could prevent infection was demonstrated with zidovudine, which protected infant macaques following a low-dose intravenous inoculation. 152 Subsequently, a growing series of studies that used lower virus doses, sometimes combined with a mucosal route of virus inoculation, have demonstrated that administration of a number of antiretroviral drugs starting prior to, at the time of virus inoculation, or shortly thereafter was able to prevent infection at varying success rates. 60,153 –159,160 –163 Although direct comparisons were not always done, all available data indicate that (1) preexposure prophylaxis (PrEP) is more successful than postexposure prophylaxis (PEP), (2) the intravenous route of virus infection is the most difficult one to protect against, similar to what is observed in SIV vaccine studies, (3) chemoprophylactic efficacy against one mucosal route of virus inoculation can generally be extrapolated to other mucosal routes, and (4) for postexposure prophylaxis, a combination of the timing and duration of drug administration determines the success rate, as a delay in the start, a shorter duration, or interruption of the treatment regimen all reduced the prophylactic efficacy, 131,154,164 –168 (5) in some postexposure prophylaxis studies, efficacy was mediated through the induction of antiviral immune responses that controlled infection, as demonstrated by resistance against viral rechallenge or by CD8+ cell depletion experiments, 167,169 and (6) some drugs such as tenofovir can still be (partially) effective in protecting macaques following inoculation of viral mutants with a lysine-to-arginine substitution in RT (K65R) associated with reduced in vitro susceptibility to tenofovir, particularly when the mucosal route of virus inoculation was used, or when animals had some preexisting antiviral immune responses that by themselves were not protective. 170,171
In recent years, tenofovir and emtricitabine (FTC) have received growing attention for chemoprophylaxis for several reasons. First, unlike many other compounds that showed prophylactic efficacy in nonhuman primates but are no longer in clinical development, both tenofovir and emtricitabine have been approved to treat HIV-infected people. Second, from 1995 onward, an abundance of studies in macaques has demonstrated that tenofovir alone or in combination with emtricitabine had prophylactic efficacy against all tested routes of virus inoculation (intravenous, oral, intravaginal, and intrarectal). 155,159,162,168,172 Third, in an effort to simplify regimens, several studies in infant and adult macaques have demonstrated that short or intermittent PrEP regimens of tenofovir (with or without emtricitabine) that targeted the timings of viral exposure were still effective in reducing infection rates. 155,159,162,163
The abundance of evidence that antiretroviral drugs can prevent infection in macaque models has provided the scientific impetus to test similar strategies in humans in several clinical settings. Drug regimens containing in particular zidovudine and nevirapine have proven to be very effective in reducing the rate of mother-to-infant transmission of HIV, including in developing countries. 173 –175 Although the short nevirapine regimen given at the onset of labor frequently induces nevirapine resistance mutations in the mother, 176 recent studies have demonstrated that the addition of tenofovir and emtricitabine reduces the likelihood of this nevirapine resistance. 177 –180 In addition, postexposure prophylaxis is now routinely recommended after occupational (e.g., needle-stick accidents of health care workers) and nonoccupational exposures (e.g. sex or injection drug use). 181,182
Because a highly efficacious HIV vaccine has so far not been identified, there has been a growing interest in exploring PrEP regimens as an HIV prevention strategy. The promising features of tenofovir (with or without emtricitabine), as discussed above, made these compounds the leading candidates in the recently completed or currently ongoing PrEP clinical trials that investigate whether uninfected adult persons who engage in high-risk behavior will have a lower infection rate by taking an oral tablet of these compounds daily. 163 An overview of the design and timeline of these international trials that target different high-risk populations is provided elsewhere. 126 In 2010, the iPrEx trial in men who have sex with men was the first one to demonstrate that a daily regimen of tenofovir plus emtricitabine demonstrated prophylactic efficacy that correlated strongly with adherence. 6 Since then, the Partners PrEP and TDF2 trials reported that both tenofovir and tenofovir plus emtricitabine reduced the risk of HIV transmission among men and women whose primary route of HIV exposure is penile–vaginal sex. 183,184 The results of other ongoing studies are expected within the coming few years. Although the great potential of PrEP has generated a lot of enthusiasm, the challenge remains now to untangle the myriad of practical, ethical, and financial issues for wider implementation of PrEP strategies in real-world settings. 185 Meanwhile, further studies in the animal model can continue to explore unresolved gaps, such as the impact of drug resistance on PrEP efficacy. 163
Therapy: treatment of infection
Many studies in the macaque models have demonstrated that even when infection was not prevented, early drug treatment delayed or reduced the peak of acute viremia, enhanced antiviral immune responses, and delayed disease progression. 20 The earliest reports of the benefits of early therapy in SIV-infected macaques were at that time a very important proof-of-concept, because the first human trials did not demonstrate any difference in survival between immediate administration of zidovudine monotherapy and deferred initiation of therapy 186 –188 ; in hindsight, this result was likely because in these human studies, the acute viremia stage was missed and zidovudine monotherapy was rather weak during the chronic stage of HIV infection. Since then, however, the availability of more potent drugs and earlier diagnosis of infection have led to a growing body of evidence in support of early treatment for HIV-infected adults and children, not only to benefit their own health but also to drastically cut the likelihood of transmission to others. 189 –195
When antiretroviral drugs were first used to suppress acute viremia in animals and then withdrawn, the durability of its benefits depended on the virus isolate. With pathogenic env-SHIV isolates, short-term suppression of acute viremia usually induced strong antiviral immune responses that controlled virus replication and delayed disease for an extended time in the absence of drug treatment. 196 –198 In contrast, when drug treatment was withdrawn from animals infected with virulent SIV isolates (such as SIVmac251) viremia generally increased, similar to what is commonly observed in HIV-infected humans. 66,67,199 –201
When macaques were started on antiretroviral drug monotherapy during chronic infection, the results were variable and generally less impressive than those of early treatment, as the reduction in viremia was often transient or even absent. 60,62,187,198,202 –205
Many studies demonstrated that tenofovir was generally effective in suppressing viremia during established SIV infection. 59,62,172,206 –208 However, in some studies, tenofovir therapy was not effective in suppressing viremia despite the presence of drug-susceptible virus at the onset of treatment; such poor virologic response was especially observed when tenofovir therapy was initiated when viremia was high and animals were immunosuppressed. 197,206,208 –211 Such observations indicated that the success of antiviral drug therapy is more than just a combination of sufficient drug levels and susceptible virus, and have led to the development of a model of viral dynamics during drug therapy that also incorporates the role of the immune system (see further).
With the availability of antiretroviral drugs that can suppress virus replication in infected macaques, these drugs can now be combined to achieve better suppression of virus replication and reduce the likelihood of the emergence of drug-resistant mutants, similarly to ART in humans; such combination regimens commonly include tenofovir and emtricitabine. 201 Antiretroviral drugs can now also be used as tools to gain deeper insights into disease pathogenesis and drug therapy. Examples of such studies include the use of antiretroviral drugs to do mathematical modeling, to study the role of CD8+ cell-mediated immunity during SIV infection, and to test the effects of early antiretroviral therapy on gut-associated lymphoid tissue (GALT) and viral reservoirs. 212 –218
Many studies have combined antiretroviral drugs with immunotherapeutic strategies with the aim of restoring the immune system or enhancing antiviral immune responses so that when drug treatment was stopped, viremia was controlled better. These strategies included structured treatment interruption, and a variety of active immunization strategies. 199,219 –234 While most of these studies provided proof-of-concept of efficacy, the benefits were usually variable or transient, or the strategy was technically challenging and would first require simplification to make it applicable on a large scale. Accordingly, the current status—as of 2011—is that sustained suppression of viremia in SIV-infected macaques or HIV-infected humans to undetectable or very low levels generally still requires daily administration of antiretroviral drugs. 132,235
The role of primate models in studying drug resistance
ART successfully suppresses virus replication in many but not all HIV-infected individuals. While other factors such as compliance and pharmacokinetics also contribute to reduced efficacy of ART, a major limiting factor is the emergence of viral mutants with reduced in vitro susceptibility to antiviral drugs. 236,237
While the correlation between specific mutations in the viral genome and in vitro reduced susceptibility has been documented extensively for many antiviral compounds, the terms “drug resistance” and “reduced susceptibility” remain in vitro measures. Many unanswered questions remain regarding the in vivo virulence and exact clinical implications of these drug-resistant variants in vivo, and, thus, how to use such information to make treatment decisions (Fig. 4).
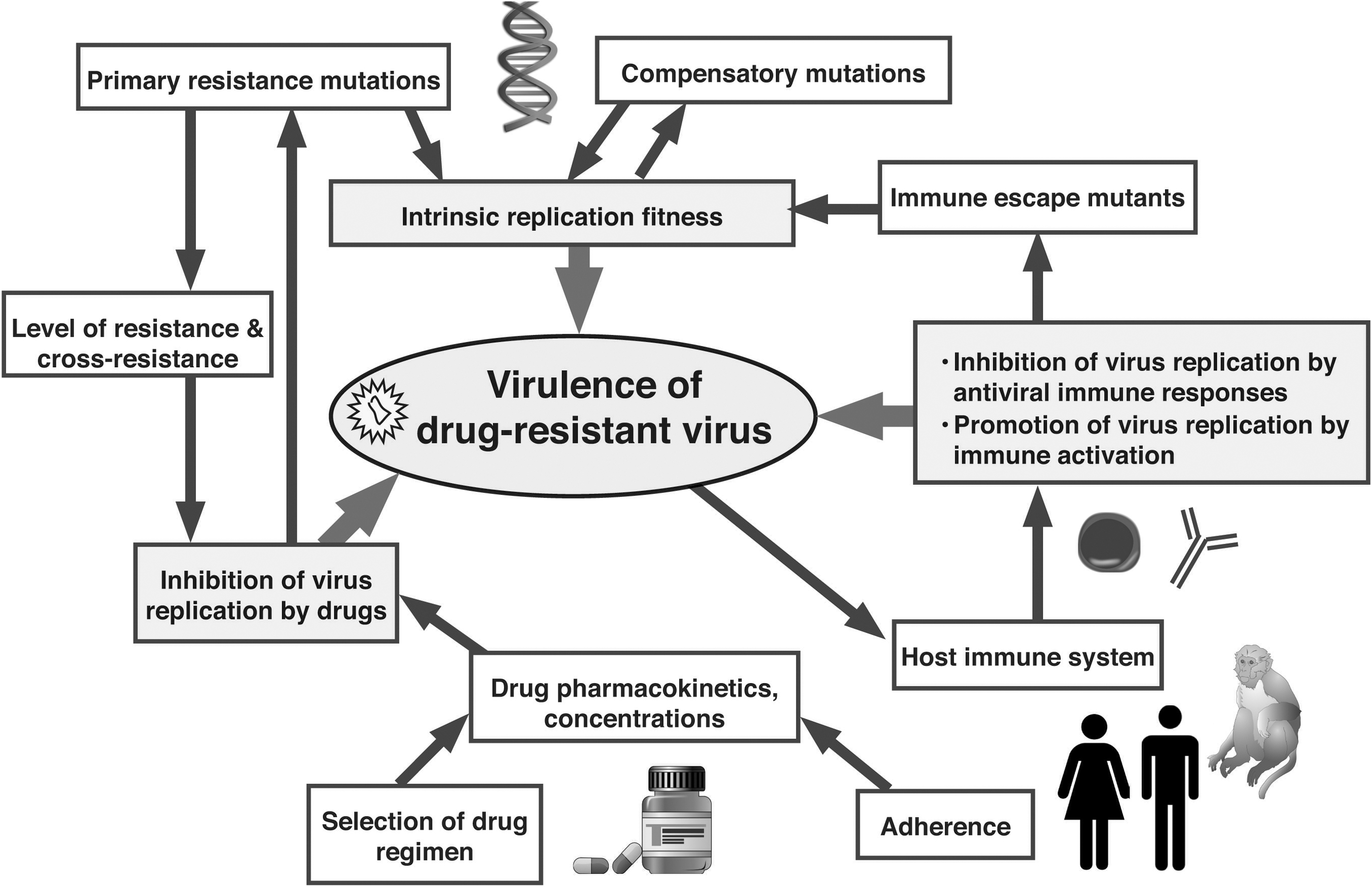
Determinants of in vivo virulence of drug-resistant viral mutants. The ability of drug-resistant virus to cause disease is determined by a complex interplay of many viral, host, and pharmacologic forces. Antiviral drugs and antiviral immune responses select for the emergence of drug-resistant and immune escape mutants, respectively. As such mutations are often associated with a reduction in viral replication fitness, compensatory mutations may develop in an attempt to restore this viral fitness. Although the immune system tries to control virus replication, the resulting immune activation can promote virus replication and contribute to the demise of the immune system. As supported by data from animal models and human studies, a minor change in one of these forces (e.g., viral fitness, or residual antiviral drug activity) can have a major impact on the overall outcome if it provides an additional opportunity for another arm (e.g., the immune system) to helping reducing viral replication.
Macaque models have been used to gain insights into the emergence and clinical implications of such drug-resistant mutants. Several studies have described the emergence of drug-resistant viral mutants in SIV- or SHIV-infected macaques during prolonged treatment with zidovudine, nevirapine, efavirenz, lamivudine (3TC), emtricitabine (FTC), or tenofovir; in general, the mutational patterns found in SIV or RT-SHIV were consistent with those found in HIV-1- or HIV-2-infected people who received these drugs. 203,238
Although the emergence of drug-resistant mutants in drug-treated macaques is an important first step, the extra value of the animal models lies in their ability to help unravel the clinical implications of such viral mutants. A first question about drug-resistant mutants concerns their in vivo replicative fitness and virulence in comparison to wild-type virus. A second one is how they affect the response to treatment: if drug resistance means that the drug is no longer effective, then the compound may as well be withdrawn, but if there is still a partial response, then it will be wrong to discontinue drug administration unless better therapies can be offered.
Some information on the relative replication fitness, stability, and clinical implications of drug-resistant HIV mutants in vivo can be gathered from case reports that document primary infection with drug-resistant HIV-1, as well as studies that measured the impact of treatment interruption on viremia and the reversion of drug-resistant virus to wild-type. 239 –242 In contrast to what is feasible in humans, an animal model allows the most direct and controlled approach to study the replication fitness, virulence, and clinical implications of drug-resistant virus: animals can be inoculated with drug-resistant viral mutants or their wild-type counterparts, and viremia and clinical outcome can be compared in drug-treated versus untreated animals.
A first study demonstrated that the zidovudine-induced Q151M RT mutation in SIVmac251, which is the result of two base changes, was stable in the absence of zidovudine treatment and did not significantly reduce viral replication fitness and viral virulence. 243 In another study, the lamivudine- and emtricitabine-selected M184V mutation in RT was found to slightly impair the in vivo replication fitness of SIVmac239, but this minor impairment was not sufficient to affect viral virulence, because the disease course in animals inoculated with M184V virus and treated with emtricitabine (to prevent reversion) was indistinguishable from that of untreated animals infected with wild-type virus. 203
The SIV and RT-SHIV macaque models have also been used to investigate the clinical implications of K65R RT mutants, which have a 5-fold reduced in vitro susceptibility, during prolonged tenofovir treatment. Following the emergence of K65R mutants, two outcomes were observed during continued tenofovir therapy: (1) viremia remained or became high, but continued tenofovir treatment still improved survival more than predicted by viral RNA levels and CD4+ T cell counts; or (2) K65R viremia was reduced and could remain very low or undetectable with prolonged disease-free survival (>3–14 years). 59,132,209,211,244,245 To further explore these observations, K65R SIV isolates were inoculated into new animals. In the absence of tenofovir treatment, K65R SIV isolates were as fit and virulent as wild-type virus. 245 In contrast, in the presence of tenofovir treatment, the disease course of K65R virus-infected animals was delayed. 245 In the tenofovir-treated animals with low viremia of K65R viral mutants, withdrawal of tenofovir therapy generally led to a slow increase in viremia. 209,244 While these observations suggest residual antiviral effects of tenofovir against these mutants, the immune system was also found to play a major role in suppressing K65R viremia (see the next section).
In summary, these studies with antiretroviral drugs in macaques, which generally used monotherapy, demonstrate the concept that depending on the drug and the individual animal, the clinical implications of drug-resistant virus can vary a lot: it can range from loss of benefits to sustained benefits of antiretroviral therapy. Although the situation in HIV-infected humans—where a myriad of drug resistance mutations can accumulate after exposure to many antiretroviral drugs—is much more complex, there is now also a growing body of evidence, often derived from drug interruption studies, that certain drug regimens can still provide therapeutic virologic and/or immunologic benefits in the presence of drug-resistant virus, due to some residual activity of drugs against the resistant variant and the selective maintenance of a virus with reduced replicative capacity. The available data suggest that the roles of these mechanisms differ according to the therapeutic drug class. 242,246 –252 Such information is especially relevant for heavily drug-experienced HIV-infected people with persistent viremia, for whom treatment options are often limited and for whom their clinicians have the dilemma of considering “when to switch,” “how to switch,” and “how to wait.” 251 However, because the ultimate goal of ART remains the maximal suppression of viremia and to indefinitely halt disease progression, this emphasizes the continued need to develop novel compounds or novel combination regimens that reduce the likelihood of resistance and bear the least cross-resistance with current regimens.
The role of the immune system on the efficacy of drug therapy
For many years, relatively little attention was given to the role of antiviral immune responses during effective drug therapy, because antiviral immune responses were mostly regarded as a backup plan to contain viremia whenever drug treatment was withdrawn or drug-resistant virus would emerge. 253 In recent years, however, a growing body of evidence from both primate and human studies demonstrates that antiviral immune responses play a more important role during drug therapy. 198,211,244,254,255 The current review will briefly highlight various observations from macaque studies that support the development of a model that incorporates how antiviral immune responses can work in synergy with antiretroviral drugs.
First, several studies have indicated that the virological response of SIV- or RT-SHIV-infected macaques following initiation of antiretroviral therapy depended on their major histocompatibility complex (MHC) alleles, which suggested a role of the immune system. 209,256,257
Additional support for the role of immune responses is provided by reports that drug regimens that were very effective in suppressing viremia in macaques during acute viremia became less effective when therapy was started later in infection, even though the virus had wild-type susceptibility at the onset of treatment; this was mostly observed when more virulent SIV and SHIV isolates were used, which induce high viremia and severe immunodeficiency. 197,198,206,208,210,211 These findings are consistent with observations in humans, where lower CD4+ cell counts and higher pretreatment virus levels have generally also been associated with a slower virological response to therapy. 258 –260
An advantage of animal models is that it allows approaches that are not feasible in humans, such as selective depletion of CD8+ cells via administration of monoclonal antibodies. Such CD8+ cell depletion studies in tenofovir-treated macaques have demonstrated the concept that tenofovir therapy required the assistance of immune responses to reach full effectiveness in reducing viremia, both at the onset of treatment when the virus has wild-type susceptibility, as well as during prolonged treatment in the presence of drug-resistant K65R mutants. 20,209,244 Tenofovir-treated animals that have K65R viral mutants are able to maintain very low viremia for many years due to this combination of strong antiviral immune responses and continued therapy, because either CD8+ cell depletion or temporary interruption of tenofovir therapy resulted in a transient increase of viremia. 132,209,244 A similar synergistic effect was also observed in a prophylaxis study where both antiviral immune responses (induced by prior immunization) and short-term tenofovir treatment were required to protect macaques against infection after intravenous inoculation with a high dose of a virulent K65R SIV isolate. 170
These data led to the development of a model, described in more detail elsewhere, in which both antiviral drugs and antiviral immune responses are required to obtain a maximal and sustained suppression of viral replication of wild-type virus, as well as of drug-resistant viral mutants. 20 In this model of drug therapy, antiviral immune responses contribute significantly to the antiviral efficacy of drugs by reducing the burst of virus replication in productively infected cells via cytolytic or noncytolytic pathways. In the absence of such antiviral immune responses, antiviral drugs face a more daunting task to control viremia as already infected cells can survive longer and produce more viral progeny. 20,217,218
This model can also include the effects of altered replication fitness or diversity of mutant virus, and/or residual drug activity (Fig. 4). In particular, even a relatively minor decrease in replication fitness or viral heterogeneity, or a partial inhibition of virus replication by the drug regimen, can have a major impact on viremia if it provides more opportunity for effective antiviral immune responses to kill or inhibit productively infected cells prior to the major viral burst; in contrast, without effective antiviral immune responses (such as during late-stage disease), a small difference in replication fitness or residual drug activity may not translate into any significant difference in viremia and clinical outcome. 203,244,261
Incorporating the role of the host immune system in the way we interpret viral dynamics during drug therapy is useful for several reasons. First, the variability in the strength of antiviral immune responses may be one of the contributing factors that explain the variability in the virologic responses that are observed among individuals and among study cohorts. 262 –264 The demonstration that a combination of tenofovir and antiviral immune responses can suppress K65R SIV replication in macaques for many years is also consistent with observations that viremia in persons with detectable K65R mutants can be suppressed by tenofovir-containing regimens, and it also explains observations of strong antiviral immune responses in some HIV-1-infected people who are receiving ART and have low-level viremia with drug-resistant virus. 255,265 –267 Finally, all these observations spark cross-fertilization of the research agendas of pharmacology and immunology. They provide a strong scientific rationale for a continued exploration of immunotherapeutic strategies that induce antiviral immune responses that in concerted action with antiretroviral drugs are more likely to indefinitely halt disease progression.
A Glimpse into the Future: The Ultimate Goal of Stopping ART
Considering the bleak prognosis for HIV-infected patients during the early years of the epidemic, our current ability to manage HIV infection with antiretroviral drugs and other supportive interventions represents a triumph of research and modern medicine. 7 However, because even the newer drugs are still relatively expensive and carry some risk of toxicity (which may be cumulative after decades of treatment), the ultimate dream remains to discover a strategy that would allow permanent withdrawal of ART, either by achieving long-term immunological control (as discussed earlier) or by purging out all viral reservoirs and totally curing HIV infection. The observation that HIV-1 infection seems to have been cured in the so-called Berlin patient by irradiation (to treat leukemia) followed by a bone marrow transplant from a donor with the Δ32 CCR5 mutation provided proof-of-concept and has initiated a “race for the cure.” 268
Although early mathematical models predicted that several years of ART may perhaps be able to cure infection, 269 the much slower decay rates of some viral reservoir compartments and/or evidence of some ongoing low-level virus replication have dashed such hopes, because treatment interruptions invariably result in a rebound of virus replication. 270 Thus, novel strategies have to be explored. The available evidence suggests that viral persistence during ART can be caused by different mechanisms, of which the relative importance is still being debated, and which may vary depending on the drug regimen, viral factors, and host factors. Thus, we are faced with the daunting task that most likely all of these mechanisms need to be targeted simultaneously to be successful. 271 First, evidence of very low levels of virus replication during ART with ongoing replenishment of the reservoirs suggests the need to develop even more effective antiretroviral drug regimens (e.g., drugs that target the regulatory and accessory proteins of HIV) or gene therapy-based approaches that inhibit virus replication. 272,273 Second, to reduce the latent viral reservoirs, agents are required to activate HIV gene expression. 12 Third, because viral gene expression and viral replication by themselves may not result in the death of the infected cell, strategies are needed to selectively and effectively destroy the HIV-infected cells in the reservoirs. 271
Many strategies aimed at reactivating latent virus and eradicating viral reservoirs are likely to carry the risk of adverse effects such as toxicity or, in the case of incomplete eradication, a rebound of viremia upon withdrawal of ART, which may affect future treatment options. Without proof of concept, only a few people who fare well on a stable ART regimen may be interested in volunteering for such clinical trials. Because animal models have been improved due to the availability of better drugs and more sensitive methods to detect virus, they are likely to play a crucial role in better understanding viral persistence and testing novel strategies that target the different mechanisms of viral persistence with the aim of curing infection. 12
Studies to reactivate latent virus and reduce viral reservoirs have been initiated in murine and nonhuman primate models, and some studies demonstrated an effect on viral rebound upon drug withdrawal. 274 –278 Because ART treatment of SIV-infected nonhuman primates also results in undetectable or very low viremia and the establishment of viral reservoirs, including resting T cells, this animal model is also suitable to further explore such strategies. 212,214
In summary, although the ultimate goal of eradicating infection is currently still distant, reaching it will most likely depend on continued progress in our understanding of SIV/HIV pathogenesis; the steps that are currently being taken are, albeit relatively small, important and crucial to guide us in the right direction.
Conclusions
During the past three decades, since the initial discovery of HIV and SIV, the nonhuman primate models of HIV infection have evolved dramatically. Despite many initial obstacles, the gradual development of better resources (reagents, assays, and drugs) and our better understanding of disease pathogenesis have improved the usefulness of nonhuman primate models to evaluate novel prophylactic and therapeutic drug strategies, and to address hypotheses that cannot be tested appropriately by in vitro experiments and are difficult to explore in humans.
The relevance of the model has been demonstrated by several antiviral strategies that, after first having been discovered in nonhuman primates, were able to enter the pipeline of clinical trials and are now increasingly used in humans. Nonhuman primate models are also likely to play a crucial role in the development of strategies aimed at reducing viral reservoirs and ultimately curing infection.
Although each of the available models has its intrinsic limitations and advantages, ongoing comparison of results obtained in nonhuman primate models with those observed in human studies will lead to further validation and improvement of these animal models so they can continue to advance our scientific knowledge and guide clinical trials.
Footnotes
Acknowledgments
We thank Dr. M. Marthas for a critical review of the manuscript and Kathy West for assistance with the illustrations.
Author Disclosure Statement
No competing financial interests exist.