
Abstract
HIV/tuberculosis (HIV/TB)-coinfected patients intolerant/resistant to nonnucleoside reverse transcriptase inhibitors (NNRTIs) have limited treatment options. We evaluated the pharmacokinetics (PK)/safety/efficacy of an adjusted dose of indinavir/ritonavir (IDV/r) 600/100 mg plus two NRTIs in HIV/TB-coinfected Thais receiving rifampicin-based anti-TB treatment. This was a prospective, open-label study. Eighteen Thai, HIV/TB-coinfected patients between 18 and 60 years were recruited. IDV/r 600 mg/100 mg plus lamivudine and stavudine were administered every 12 h (bid). When rifampicin was stopped, IDV/r was reduced to 400/100 mg BID. Clinical outcomes, adverse events, and concomitant drugs were intensively collected. Intensive 12-h PK was performed after 2 weeks of IDV/r while on rifampicin. Samples were collected: predosing and 1, 2, 3, 4, 6, 8, 10, and 12 h after drug intake. The median body weight was 55 kg. The median CD4 was 26 cells/μl. The median HIV RNA was 5.05 log10 copies/ml. Then 15/18 underwent intensive PK at week 2. The median time between initiating rifampicin and IDV/r was 4.5 months. The median duration of rifampicin during study (rifampicin/IDV/r together) was 15.6 weeks. All received a total of 9 months of antituberculous drugs. The geometric means (GM) of indinavir AUC0–12 and C 12 were 8.11 mg*h/liter and 0.03 mg/liter, respectively. After stopping rifampicin and reducing IDV/r to 400/100 bid, the GM indinavir C 12 increased to 0.68 mg/liter (p=0.004). In all, 8/18 (44%) had asymptomatic ALT elevation and 2/18 (11%) had symptomatic hepatotoxicity requiring IDV/r discontinuation. All 13 patients who remained on IDV/r treatment had HIV RNA <50 copies/ml at 48 weeks. Concomitant use of rifampicin and IDV/r resulted in subtherapeutic indinavir concentrations. Although 44% of them developed asymptomatic Grade 3/4 transaminitis, the rate of study drug discontinuation due to hepatotoxicity was low. Despite good virological outcome in our cohort, prolonged exposure to subtherapeutic indinavir concentrations may lead to treatment failure.
Introduction
C
In Thailand, tuberculosis accounts for about 30% of HIV-infected patients. 1 Despite the availability of effective therapy for both diseases, simultaneous treatment continues to be one of the most complex scenarios due to drug–drug interactions, high pill burden, immune recovery syndrome, and overlapping toxicities. 2,3 Based on the SAPiT trial, 4 initiating HAART during TB treatment reduces all-cause mortality by 56% in sputum smear-positive TB-HIV-coinfected patients with CD4 <500 cells/μl. This trial provides strong justification for initiating HAART at some point during the course of TB treatment and not deferring HAART until after the completion of TB treatment in the HAART-eligible patient.
In regards to TB treatment, rifampicin is the cornerstone of effective anti-TB treatment because it is more effective than non-rifampicin-containing regimens and allows for shorter courses of therapy with higher rates of treatment success. 5 –7 Unfortunately, rifampicin is a potent inducer of hepatic cytochrome P450 3A (CYP3A) and drug transporter P-glycoprotein, 8 –10 which results in markedly lower plasma concentrations of boosted PIs such as indinavir/ritonavir, 11 atazanavir/ritonavir, 12 –14 saquinavir/ritonavir, 15 –18 and lopinavir/ritonavir. 19 Several studies in healthy volunteers have attempted to overcome this by using higher doses of boosted PI; however, high rates of liver toxicity were seen. 9,10,18 –20 WHO 21 and the U.S. Department of Health and Human Services 22,23 therefore recommend refraining from using a boosted PI with rifampicin until more data are available. These guidelines instead recommend rifabutin, a weaker inducer of CYP3A that appears to cause less pronounced reductions in boosted PI exposure than rifampicin. 24 Although rifabutin has recently been added to the WHO Essential Medicines List, it is not available in Thailand and in many other countries. In addition, data supporting rifabutin efficacy and safety in HIV-infected patients are scarce 25 and when used with boosted PI, dose adjustment of both drugs is required. 26 Therefore, there is an urgent need to study the pharmacokinetics (PK) of boosted PI and rifampicin in HIV/TB patients. In Thailand, indinavir/ritonavir (IDV/r)-based HAART was the most used boosted PI regimen for the national program until recently when lopinavir/ritonavir (LPV/r) became more widely available. Even though indinavir is no longer the preferred option, however, it is still used in some patients. Indinavir/ritonavir 400/100 mg twice daily is the recommended dose for Thai patients. 27,28 To obtain a higher indinavir plasma concentration, we increased the dose of boosted indinavir and hoped that the renal functions would remain safe. In this study, we evaluated the pharmacokinetics of an adjusted dose of IDV/r in HIV/TB patients before and after rifampicin-based TB treatment.
Materials and Methods
This was a prospective, open-label study with a total of 18 Thai patients concurrently infected with HIV-1 and TB. Subjects were recruited through the HIV-Netherlands Australia Thailand Research Collaboration (HIV-NAT), The Thai Red Cross AIDS Research Centre, and King Chulalongkorn Memorial Hospital, Bangkok, Thailand. HIV-1-infected men and women between 18 and 60 years old were enrolled if they also had active TB and had been taking rifampicin-containing anti-TB treatment for at least 2 weeks. TB was diagnosed by clinical symptoms plus acid fast stain and/or positive culture for Mycobacterium tuberculosis. All patients with CD4 >350 cells/μl, serum creatinine levels >1.4 mg/dl, and aspartate aminotransferase (AST) and alanine aminotransferase (ALT) levels >5 times the upper limit of normal range were excluded. The study was approved by the Institutional Review Boards of the Faculty of Medicine, Chulalongkorn University. All procedures were in accordance with Helsinki Declaration of 1975 (as revised in 1983). All subjects provided written informed consent before starting the study. This study was registered at clinicaltrials.gov: NCT 00411996.
At enrollment, patients commenced IDV/r 600 mg/100 mg (IDV/r) plus lamivudine 150 mg and stavudine 30 mg every 12 h with food. After 24 weeks, stavudine (d4T) was switched to zidovudine (AZT) 300 mg or 200 mg twice daily. The reason for switching from d4T to AZT was because we wanted to fade out d4T to prevent long-term d4T toxicity problems. We initially used d4T because our very advanced HIV-infected ARV-naive Thai patients had better tolerance and fewer adverse effects on it compared to AZT. IDV/r 600/100 mg twice daily was switched to IDV/r 400/100 mg twice daily (standard dose for Thais) 27,28 when patients stopped taking rifampicin. All patients received 2 months of isoniazid, rifampicin, ethambutol, and pyrazinamide and 7 months of isoniazid and rifampicin. The dosage of rifampicin was 450 mg per day for patients who weighed <50 kg and 600 mg per day for those who weighed ≥50 kg.
Patients were seen at weeks 1, 2, 4, 8, 12, 16, 20, 24, 36, and 48. Information on clinical outcomes, adverse events, drug adherence by pill count, and concomitant drugs was recorded at each time point. Blood samples were obtained to determine CD4+ cell counts (by flow cytometry), HIV viral load (HIV RNA) (by Roche Amplicor, version 1.5), and complete blood cell counts at baseline and weeks 12, 24, and 48. ALT levels were measured at baseline, days 3, 5, and 7, and weeks 2, 4, 8, 12, 16, 20, 24, 36, and 48. Intensive PK study was performed at week 2. Trough plasma concentrations of indinavir and ritonavir were obtained after 12 h of IDV/r administration (C 12 h) at week 4 and at least 4 weeks after stopping rifampicin. After completing the study, in order to follow the subjects' TB treatment outcome as well as continue care including HIV treatment, patients were enrolled in a prospective long-term cohort.
Pharmacokinetic analysis
Intensive 12-h PK study was performed after 2 weeks of IDV/r while all patients were on rifampicin. Samples were collected predosing and 1, 2, 3, 4, 6, 8, 10, and 12 h after drug intake. Samples were processed within 2 h after blood collection and centrifuged at 3000 rpm for 10 min at 20°C. All samples were frozen at less than −20°C. All subjects were observed when they took IDV/r with a standardized breakfast of 550 kcal and 12 g of fat. Other meals and snacks throughout the day were also standardized and no other food was allowed. Plasma concentrations of indinavir and ritonavir were determined by a validated high-performance liquid chromatography (HPLC) method with UV detection. 29 The indinavir level was linear over the range of 0.045–30.0 mg/liter. The percentage accuracy was found to be 102% at 0.15 mg/liter, 105% at 1.5 mg/liter, and 105% at 7.5 mg/liter. The percentages CV of within day precision were 3.22% at 0.15 mg/liter, 4.08% at 1.5 mg/liter, and 2.88% at 7.5 mg/liter, and the percentages CV of between day precision were 5.81% at 0.15 mg/liter, 0.0% at 1.5 mg/liter, and 0.0% at 7.5 mg/liter. The lower limit of quantification for indinavir was 0.045 mg/liter. For the ritonavir level, it was linear over the range of 0.045–30.0 mg/liter. The percentage accuracy was found to be 101% at 0.15 mg/liter, 104% at 1.5 mg/liter, and 103% at 7.5 mg/liter. The percentages CV of within day precision were 3.22% at 0.15 mg/liter, 1.70% at 1.5 mg/liter, and 0.89% at 7.5 mg/liter, and the percentages CV of between day precision were 3.64% at 0.15 mg/liter, 1.17% at 1.5 mg/liter, and 1.10% at 7.5 mg/liter. The lower limit of quantification for ritonavir is 0.045 mg/liter. None of our subjects had indinavir and ritonavir plasma concentrations below the limit of assay quantification. The assay was externally validated. 30
Pharmacokinetic parameters were calculated by noncompartmental methods using the WinNonlin software package (version 5.0.1; Pharsight Corporation, Mountain View, CA) and the AUC0–12 was calculated by using the log/linear trapezoidal rule.
Statistical methods
The primary outcome of interest was the 12-h intensive pharmacokinetic parameters of indinavir at steady state (area under curve: AUC0–12; maximum concentration: C max, trough concentration: C 12). The secondary endpoints included (1) proportion of subjects with undetectable HIV-1 RNA (<50 copies/ml) at weeks 12, 24, and 48, (2) median change from baseline in CD4 count (cells/μl), (3) median change in ALT, (4) proportion of subjects with ALT grade 3 or 4, (5) proportion of patients with C 12 of indinavir concentrations less than the recommended lower limit (<0.1 mg/liter), 31 and (6) development of HIV resistance mutations at week 48. All analyses were performed on an intent-to-treat basis (ITT) and on-treatment (OT) analysis using SAS, version 5.1 (SAS Institute, Inc., Cary, NC). The median, interquartile range (IQR; 25–75%) and 90% confidence intervals for AUC0–12, C max, and C 12 were calculated. Median (IQR) and frequency (%) were used to describe the patients' demographic characteristics for continuous and categorical data, respectively. All tests were two-sided and α <0.05 was considered statistically significant.
Results
A total of 18 HAART-naive subjects who were on rifampicin-based anti-TB treatment were enrolled. The median age of the subjects was 32 years (IQR 30–38 years) with a median body weight of 55 (IQR 46.0–62) kg. A total of 72% were male. The median CD4 was 26 (IQR 14–82) cells/μl and the median HIV RNA was 5.05 (IQR 4.82–5.3) log10 copies/ml. The median (IQR) time between initiation of rifampicin-based TB treatment and IDV/r-based HAART was 4.5 months (IQR 2.1–6.3 months) and all patients continued TB treatment for a total of 9 months in the study. The median (IQR) duration of rifampicin during study (rifampicin/IDV/r together) was 15.6 (6.8–22) weeks. All received a total of 9 months of antituberculous drugs. Five cases were on rifampicin dosed at 450 mg per day and another 13 cases were on rifampicin 600 mg per day. The patients' demographic characteristics are summarized in Table 1.
RIF, rifampicin; IQR, interquartile range; VL, viral load; ALT, alanine aminotransferase.
Pharmacokinetics
Fifteen patients underwent intensive pharmacokinetic analysis at week 2 while on IDV/r 600/100 mg twice daily and rifampicin (Table 2). Three patients did not participate in the PK study for the following reasons: one was lost to follow-up after the baseline visit and two had baseline rifampicin-induced cholestasis and had IDV/r discontinued during the first week. The geometric mean (90% CI) of indinavir C 12 was 0.03 (0.02–0.05) mg/liter; only one patients had a value above the presumptive minimum effective C trough concentration of 0.10 mg/liter. 23 At week 4 of combined TB and HIV drug, the geometric mean (90% CI) of indinavir C 12 was 0.08 (0.03–0.2) mg/liter. After the patients discontinued rifampicin for 4 weeks, the geometric mean (90% CI) of indinavir C 12 rose to 0.68 (0.5–0.93) mg/liter despite being on a reduced dose of IDV/r 400/100 mg twice daily. Figure 1 depicts the rise of C 12 after rifampicin was discontinued in individual patients.
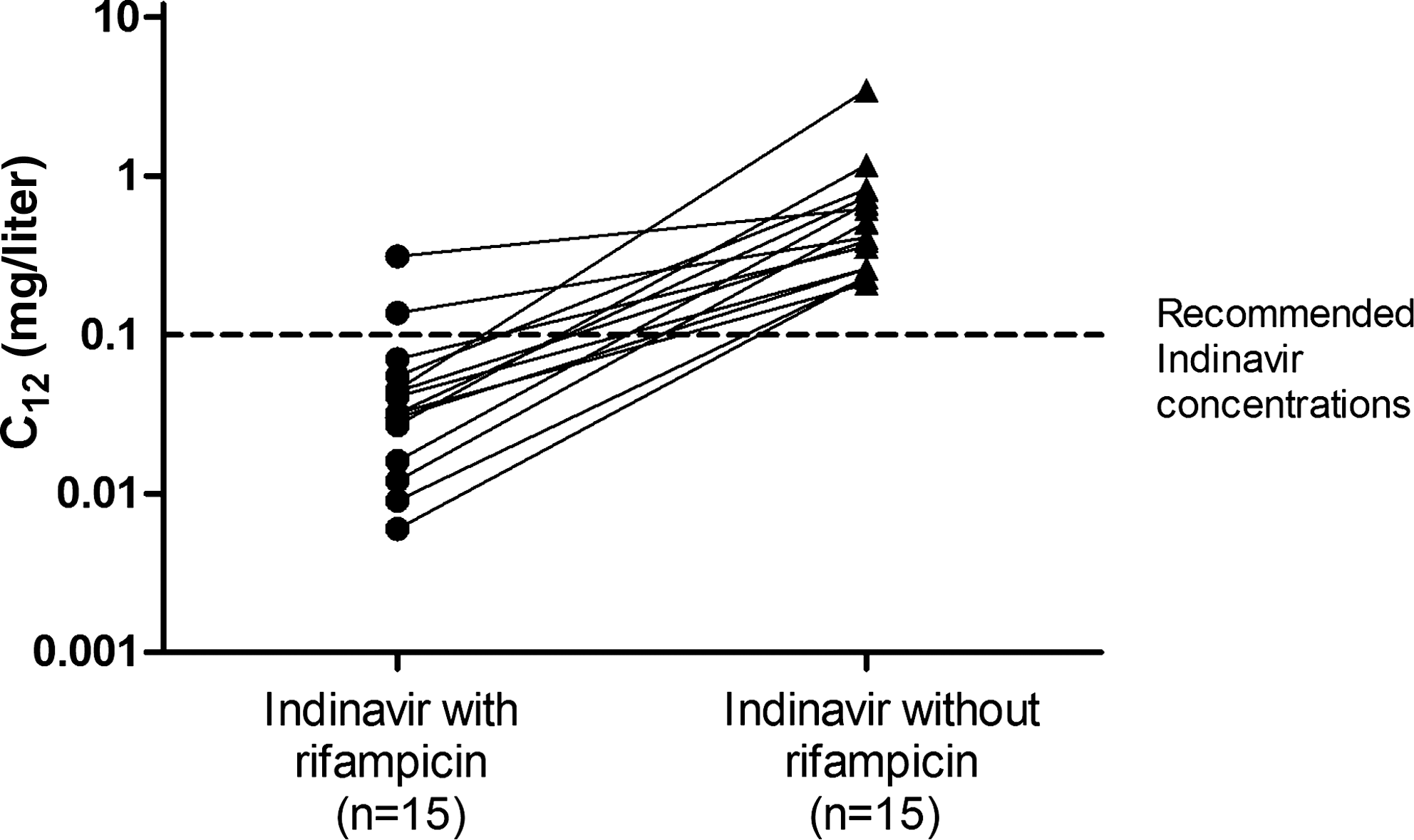
Individual indinavir C trough concentrations (C 12h) with and without rifampicin. The dosages of indinavir and ritonavir with or without rifampicin were different between both groups. Patients who took indinavir 600 mg and ritonavir 100 mg twice daily with rifampicin were compared to those who took indinavir 400 mg and ritonavir 100 mg twice daily without rifampicin. Solid lines show the median interquartile range (IQR).
Efficacy and safety
Of the 18 enrolled patients, five did not take IDV/r beyond week 24: one was lost to follow-up, two had baseline cholestasis from rifampicin, and two discontinued IDV/r at week 20 due to persistent grade 3/4 ALT elevations. For the remaining 13 patients, 92% and 100% had HIV RNA below 50 copies/ml at weeks 24 and 48, respectively. The proportions of patients achieving HIV RNA below 50 copies/ml by intention to treat analysis (n=18) were 67% and 76% at weeks 24 and 48, respectively. The median CD4 increased from 26 (IQR 14–82) cells/μl at baseline to 173 (IQR 144–282) cells/μl at week 48. All patients in follow-up completed 9 months of TB treatment. Prior to completion of TB treatment, all patients were asymptomatic and had negative AFB stain/culture. There were no TB relapses during the study.
Forty-four percent (8/18) developed asymptomatic ALT ≥100 U/liter while on concomitant rifampicin and IDV/r use with 11% (2/18) developing grade 3/4 ALT elevations and symptoms of jaundice and fatigue. ALT tended to peak at days 3 and 5, and declined spontaneously by weeks 1 and 2 (Fig. 2) except in two patients who also had hepatitis C and in these patients grade 3/4 ALT rises were seen later at week 20 resulting in discontinuation of IDV/r. Five of 15 had mild dry lips from indinavir. None of them developed renal stone/renal colic problems.

Median alanine aminotransferase (ALT) and interquartile range (IQR) over time. At week 20, two patients developed symptomatic hepatitis grade 3/4.
Discussion
The primary aim of our study was to test the concept of using ritonavir-boosted indinavir with rifampicin in patients with HIV/TB coinfection. Boosted indinavir was selected because it was the most used PI in Thailand at the time this study was conducted. In addition, PK, efficacy, and safety data of IDV/r were well documented in Thais 28,32 –37 and a case report from our institution encouraged us to formally investigate its use in treatment of HIV/TB coinfection. 38
In this study, we showed that subtherapeutic concentrations of indinavir were seen in Thais who used IDV/r 600/100 mg twice daily with rifampicin 600 mg once daily, but despite this the majority of patients achieved viral undetectability at 48 weeks. About half had asymptomatic ALT elevation with 11% having grade 3/4 ALT elevation. After rifampicin was stopped, all patients achieved adequate indinavir C 12 concentrations despite IDV/r dosing being reduced to 400/100 mg twice daily. In addition, none of them developed renal stone/renal colic problems. This was because the patients had low indinavir concentrations that have been associated with a reduced risk for developing indinavir-associated renal toxicity.
The current study demonstrates that the PK interaction between IDV/r and rifampicin is similar to other reported studies of indinavir/ritonavir, 11 atazanavir/ritonavir, 12 –14 saquinavir/ritonavir, 15 –18 and lopinavir/ritonavir, 19 which showed a reduction of PI concentrations when used with rifampicin despite increasing the PI and/or the ritonavir dose. The standard dose of IDV/r used in Thais is 400/100 mg. 27,28 Despite raising the indinavir dose by 50% to 600 mg twice daily, we could not overcome rifampicin's inducing effect on indinavir pharmacokinetics. These previously published studies were mainly done in healthy volunteers; therefore, few reported immunologic and virological outcomes with PI and rifampicin concomitant use.
Our patients had advanced HIV disease with median CD4 counts of 26 cells/μl. It was not our intention to focus on patients with low CD4 counts but the majority of our HIV/TB-coinfected patients had low CD4 counts. In this study, they achieved a remarkable increase in CD4 count and HIV viral suppression despite having subtherapeutic plasma concentrations of indinavir. In addition, the AUC0–12 of the IDV/r 600/100 mg dose with rifampicin in our study (8.11 mg*h/liter) was also much lower than observed in Thai adults taking an IDV/r 600/100 mg dose in the absence of TB treatment (39.3 mg*h/liter). 37 By intention to treat analysis, the proportion of our patients achieving HIV RNA <50 copies/ml was similar to those who received saquinavir/ritonavir and rifampicin: 62.5% 16 and 90% 18 for saquinavir/ritonavir 1600/200 mg once daily and saquinavir/ritonavir 1000/100 mg twice daily, respectively. There are no efficacy data on atazanavir/ritonavir and rifampicin, only pharmacokinetic and short-term safety data showing suboptimal atazanavir concentrations 12,14,39 and hepatitis/gastrointestinal intolerance. In addition, there are no prospective long-term efficacy data on lopinavir/ritonavir and rifampicin in adults. Only one retrospective study showed that in patients using nonadjusted dosing of lopinavir/ritonavir with rifampicin (n=23), 40 62% had undetectable HIV RNA and 17% had adverse events while 40% of patients on a recommended dose of lopinavir/ritonavir (n=5) had to prematurely stop lopinavir/ritonavir due to adverse events. Recently, a report from a South African collaborative group (n=21) 41 suggested doubling the dose of LPV/r so that the concentrations of lopinavir will be sufficient as well as tolerable, which resulted in having only two patients who developed asymptomatic hepatitis.
The favorable virological response seen in our study despite subtherapeutic plasma indinavir concentrations could possibly be explained by the short duration of combined treatment of rifampicin and indinavir/ritonavir and the presence of two active NRTIs in the regimen. Nevertheless, prolonged exposure to subtherapeutic drug concentrations could predispose patients to treatment failure. 35 Another explanation could be that the target of 0.10 mg/liter is not correct; it is mainly based on pharmacokinetic/pharmacodynamic studies with unboosted indinavir, which may not apply fully for boosted indinavir regimens.
One of the main issues prohibiting the use of a PI with rifampicin is hepatotoxicity. Two patients (11%) had symptomatic liver toxicities with grade 3/4 ALT elevation at week 20. These patients were coinfected with hepatitis C, which explains why they developed liver toxicities. Another six patients had asymptomatic ALT elevations that occurred early within the first week after adding indinavir/r to rifampicin-based TB treatment but resolved within 2 weeks. Other studies using saquinavir/ritonavir 42 and lopinavir/ritonavir 19 in HIV-uninfected volunteers showed similar rates of rifampicin/boosted PI-associated hepatotoxicity after adding boosted PIs to rifampicin, but with higher rates of toxicity-related discontinuation of the medication. The pathogenesis of rifampicin/boosted PI-associated hepatotoxicity is not well understood; additive toxicity from rifampicin, isoniazid, and boosted PI, the formation of toxic metabolites, and an immune-mediated mechanism have all been postulated. In healthy volunteer studies, 19,39,42 the rates of elevated liver enzymes seem to be higher when rifampicin was started prior to the addition of the boosted PIs whereas this toxicity was substantially less in other healthy volunteer studies when rifampicin was started after initiation of HIV drugs.
Studies in healthy volunteers strongly indicate that the sequence of introducing the PI is a factor contributing to the risk of hepatotoxicity. However, it is unclear why there is a discrepancy in the rate of developing clinical hepatotoxicity in HIV-uninfected and HIV-infected individuals. We speculate that immune-mediated injury is strongly attenuated in healthy volunteers compared to HIV-infected individuals who have much lower CD4 cell counts. Another possibility is the uptake of antituberculosis drug; the bioavailability of TB drugs is reduced in patients with advanced AIDS. 43 Therefore, the findings of hepatotoxicity from the combination treatment of rifampicin and boosted PI in healthy volunteers may not apply to all HIV-infected patients with active TB because they usually have advanced HIV disease with low CD4 cell counts.
There are limitations in our study. First, our sample size was not large enough to thoroughly evaluate the long-term efficacy of rifampicin and boosted indinavir. Second, the IDV/r dosages selected in this study may not be applicable to patients of white, African, or Latin descent, but only for Thais or Asians with small body stature. In addition, indinavir is currently not recommended in U.S. 33 and European guidelines 44 due to its long-term renal toxicity and therefore has no relevance to patients from developed countries. Last, our patients were begun on antiviral therapy near the end of TB therapy with a duration of rifampicin and boosted indinavir combination of only 16 weeks. This could explain why the outcomes of our study were excellent, as PI mutations would take time to develop. Therefore, we recommend the use of therapeutic drug monitoring to monitor PI plasma concentrations in patients with rifampicin and boosted PI if resources are available. However, if such resources are not available, boosted PIs should not be coadministered with rifampicin until clear guidelines have been established.
In conclusion, this study showed that concomitant use of IDV/r with rifampicin resulted in subtherapeutic plasma concentrations of indinavir. We showed rates of hepatotoxicity similar to those reported in HIV-uninfected volunteers. However, our study had lower rates of drug discontinuation due to hepatotoxicity. Although the majority of patients achieved viral suppression, subtherapeutic indinavir concentrations could predispose them to treatment failure, particularly if the duration of exposure is longer than in our study. Therefore, such a regimen should be used only when other options are not available and for short durations with frequent ALT and clinical follow-up particularly during the first 2 weeks. In addition, such a regimen should not be used in patients with other risks for hepatotoxicity such as chronic hepatitis B or C coinfections. In HIV/TB-coinfected patients, other alternative treatments for TB such as using second generation TB drugs 45 or using other HIV drug classes such as rifampicin with raltegravir should be explored. 46 In addition, more studies on drug interactions and treatment outcome of rifabutin and PIs and other HIV drug classes will be important to provide effective and safe treatment options for HIV/TB-coinfected patients.
Footnotes
Acknowledgments
We thank all participants and staff of HIV-NAT, Thai Red Cross AIDS. This study was presented as a poster (Poster number TUPEB144) at the 5th IAS Conference on HIV Pathogenesis, Treatment and Prevention, Cape Town, South Africa on 19–22 July 2009.
This study was funded by the Commission of Higher Education (CHE) and the Aligning Care and Prevention of HIV/AIDS with Government Decentralization to Achieve Coverage and Impact: ACHIEVED Project (The Global fund Project, Thailand).
The clinical trial registration number is NCT 00411996.
Author Disclosure Statement
J.A. has received educational grants, travel grants, and/or speakers' honoraria from Roche, Gilead, Abbott, and Tibotec. K.R. has received research grants/funding, honoraria, or is a consultant or advisor to, or lecture sponsorships from Abbott, Boehringer-Ingelheim, Bristol-Myers-Squibb, Gilead, GlaxoSmithKline, Hoffmann-LaRoche, Janssen-Cilag, Merck Sharpe & Dohme, Tibotec, and Virco. M.B. has received honoraria for HIV advisory board services and presentations provided for Abbott, Bristol-Myers-Squibb, Janssen-Cilag, and Merck.