
Abstract
Activating transcription factor 4 (ATF4) is a central factor in the cellular response to multiple stresses, including altered metabolic conditions, anoxia and hypoxia, and redox stress. ATF4 is triggered by endoplasmic reticulum stress and consequent unfolded protein response. This report identifies for the first time ATF4 as a transcription factor upregulated by HIV-1 infection. Upregulation of ATF4 enhances HIV replication, by synergistic interactions with HIV Tat. Moreover, in specific cell lines ATF4 has a direct transactivating potential on the LTR, even in the absence of Tat. We also provide evidence that expression of ATF4 induces HIV reactivation in chronically infected cell lines. These results show for the first time that ATF4 induction might have an important role in HIV replication, and suggest that ATF4 might represent a convergent signaling molecule for different stressors important in regulating the HIV-1 cycle.
H
Several viral infections affect the ER, causing protein misfolding and UPR, including hepatitis C virus, 9 respiratory syncytial virus, 10 Japanese encephalitis virus, 11 adenoviruses, 12 and human cytomegalovirus. 13,14 Viruses that induce ER stress are confronted with the consequences of UPR activation, with potential detrimental effects on viral replication, such as attenuation of translation. However, ATF4 activation can be advantageous to viral infection, and some viruses modulate the UPR. During viral infection, several kinases, activated by dsRNA and ER stress, phosphorylate elF2α and ultimately increase ATF4. 15 This mechanism prevents attenuation of translation, inhibiting harmful effects of ER stress while maintaining those that may be beneficial to the virus. Furthermore, ATF4 has recently been shown to suppress IRF7 activation, inhibiting interferon (IFN) induction 16 and facilitating viral replication. For example, reovirus replication requires ATF4, 17 and influenza virus replicates less efficiently in the absence of ATF4 expression. 18 Human cytomegalovirus (HCMV) infection increases ATF4 translation, 13 and Epstein–Barr virus (EBV) latent membrane protein 1 (LMP1) activates PERK to induce phosphorylation of elF2α, which upregulates ATF4. 19 Recently, we discovered that human herpesvirus 8 (HHV-8) also induces ATF4 expression, which, in turn, enhances virus replication and promotes virus-induced angiogenesis (Caselli et al., unpublished observations). HHV-8 is frequently detected in HIV patients, where it is causally associated to the development of Kaposi's sarcoma, one of the most frequent neoplasm during AIDS. In vitro, HHV-8 transactivates HIV-1 LTR, 20 leading to increased HIV replication and reactivation from latently infected cells. 21
ATF/CREB proteins have unique binding sites in the modulatory region of the HIV-1 LTR, 4 and ATF4 cooperates with Tax protein of the human T cell leukemia virus (HTLV-1), in LTR activation. 22,23 It has also been reported that HIV Vpu prevents proteosomal degradation of ATF4, resulting in a functional overexpression of this transcription factor. 24
To elucidate the possible functional relevance of ATF4 in the replication of HIV-1, we analyzed whether ATF4 expression is upregulated in the course of in vitro HIV infection. CD4+ Jurkat T cells were infected with the lymphotropic strain HIV-1IIIB, grown and titrated as previously described, 21 at a moi of 1:1,000. Following overnight absorption, infected cells were washed and seeded 5×105/ml/well in 12-well plates. ATF4 expression was analyzed at 8, 24, and 48 hours postinfection (hpi) by real-time quantitative PCR after retrotranscription (RT-qPCR), using a specific Taqman Assay (Applied Biosystems, Foster City, CA). Amplifications were carried out in triplicate on a 7300 real time PCR system (Applied Biosystems), using an amount of cDNA corresponding to 200 ng of total RNA. Relative amounts of ATF4 and RNaseP mRNA (used as an internal control) were determined by the 2-ΔΔCt method, with the Relative Quantitation Study software (Applied Biosystems), and levels of ATF4 mRNA were expressed as fold difference compared to controls.
The results show that HIV infection induces a 9.1-fold increase of ATF4 transcription as early as 8 hpi (Fig. 1A). The increase peaked at 24 hpi, when levels of ATF4 mRNA were about 13-fold compared to controls, and persisted at 48 hpi, although ATF4 transcripts were slightly declining (8-fold compared to controls). Results were highly statistically significant, as evaluated by Student's t test (p<0.001). ATF4 transcriptional increase was accompanied by an increased protein levels, detected by Western blot. One hundred micrograms of cellular protein lysate from infected or control cells was separated by SDS-PAGE, transferred onto nitrocellulose paper (transfer buffer 25 mM Tris, 192 mM glycine, and 20% methanol), blocked for 1 h with saturation buffer (5% dehydrated nonfat milk in 10 mM Tris-HCl, pH 7.4, 150 mM NaCl, 0.1% Tween-20), and then incubated with a rabbit polyclonal antibody directed against ATF4-CREB2 (Santa Cruz Biotechnology, Inc.). Blots were then incubated with an HRP-conjugated antirabbit antibody (Santa Cruz Biotechnology, Inc.) and developed with a chemiluminescent HRP substrate (SuperSignal West Pico Chemiluminescent Substrate, Pierce). As shown in Fig. 1B, HIV infection resulted in a prompt increase of ATF4 translation, indicated by ATF4 protein accumulation at 8, 24, and 48 hpi, with a mean increase of 9.5-fold compared to control samples (p<0.005). The slight increase in ATF4 protein observed at 48 hpi was likely due to culture conditions and was not seen at later times pi (not shown).
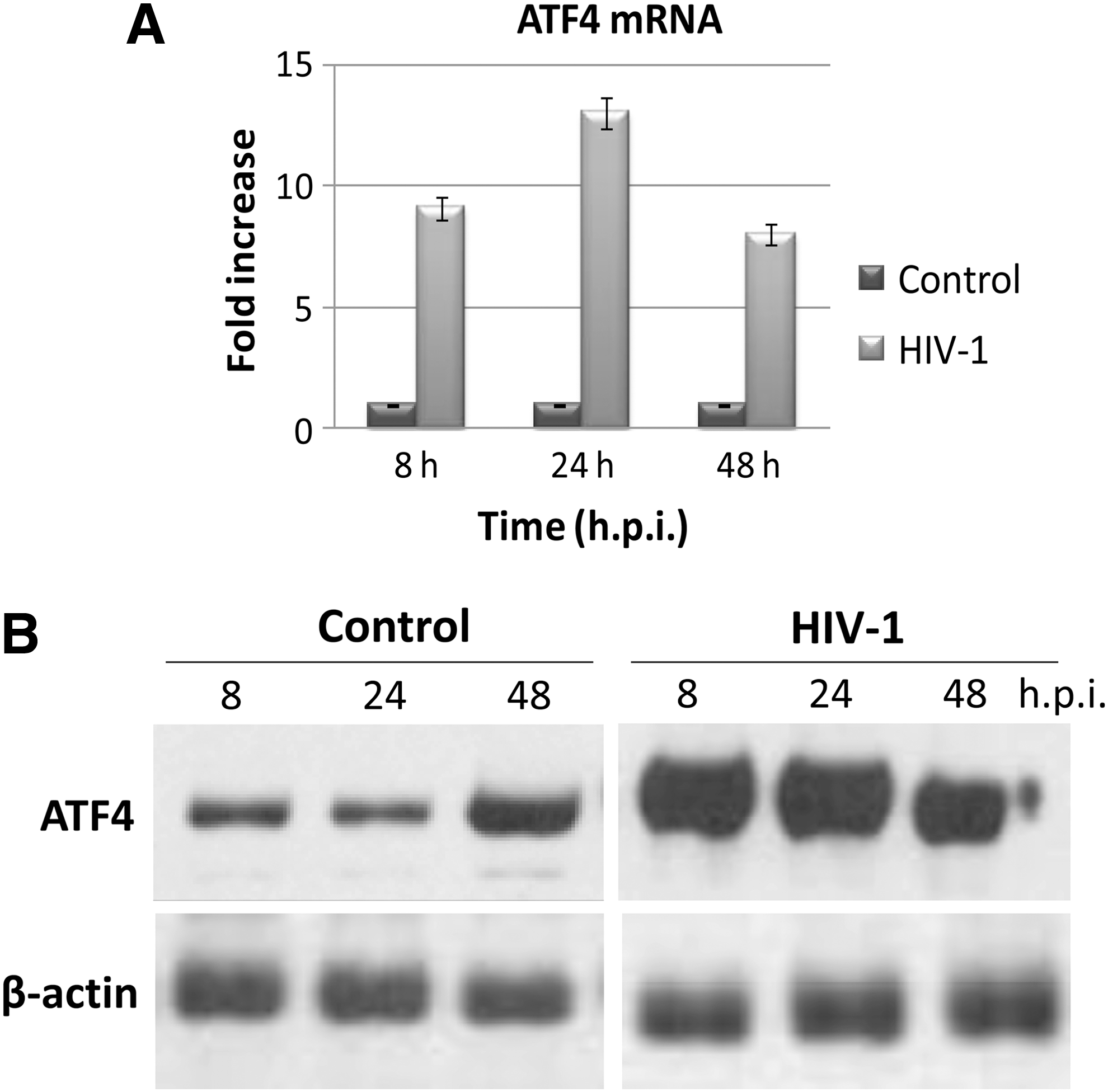
HIV-1 infection increases ATF4 expression. CD4+ Jurkat T cells were infected with HIV-1IIIB (moi 1:1000) or uninfected (controls). Samples were collected at the indicated hours postinfection (hpi).
Next, we determined whether the observed ATF4 increase has an enhancing effect on HIV replication. Jurkat T cells were transfected with pCG-ATF4, a plasmid encoding the full length ATF4 gene, 25 and subsequently infected with HIV. Control cells were untreated or transfected with pCG empty vector alone. pCG-ATF4 transfection resulted in about a 100-fold increase of ATF4 mRNA at 24 h posttransfection (hpt) (not shown), thus infection with HIV-1 was performed at 24 hpt, with a 1:10,000 moi. HIV replication was analyzed from 1 to 14 days postinfection (dpi). The presence of HIV-1 proviral DNA was analyzed by qPCR performed on total DNA, extracted as described. 21
Reactions were performed using 100 ng of total DNA and amplifying HIV-1 gag gene with the following set of primers/probe: gag(+) (5′-ATC AAG CAG CCA TGC AAA TGT T-3′); gag(-) (5′-CTG AAG GGT ACT AGT AGT TCC TGC TAT ATC-3′); gag probe (5′-FAM-ACC ATC AAT GAG GAA GCT GCA GAA TGG GA-TAMRA-3′). The standard curve was generated by amplification of serial dilutions of a plasmid (pCR-gag) containing a cloned fragment of HIV proviral DNA (gag, nucleotides 47127 to 47556), and RNaseP was used as the internal control. The results show that HIV proviral DNA was consistently more abundant in pCG-ATF4-transfected cells compared to controls (Fig. 2A). Differences, already detectable at 1 dpi, were higher at later times postinfection, when proviral genomes were up to 160-fold higher than in controls (14 dpi). Differences were statistically significant at all times tested (p<0.001). Due to the perturbation caused by nucleofection, cells receiving the empty pCG plasmid also showed a slight increase of HIV DNA, but only within a 2-fold range compared to control nontransfected cells. ATF4-transfected cells were also analyzed for HIV-1 antigen expression by immunofluorescence. Briefly, 5×104 cells were fixed with cold 4% paraformaldehyde, permeabilized with 0.25% Triton X-100, and saturated with 5% BSA in PBS for 1 h at room temperature. Slides were incubated with a mouse monoclonal anti-HIV-1 p24 antibody (Santa Cruz Biotechnology, Inc.), and secondarily incubated with an antimouse IgG (Alexa Fluor 488 conjugated, Invitrogen). ATF4-transfected cells displayed a higher expression of HIV-1 p24 antigen compared to controls, accompanied by an earlier appearance of HIV-induced syncytia, confirming that ATF4 overexpression markedly increases HIV replication (Fig. 2B).
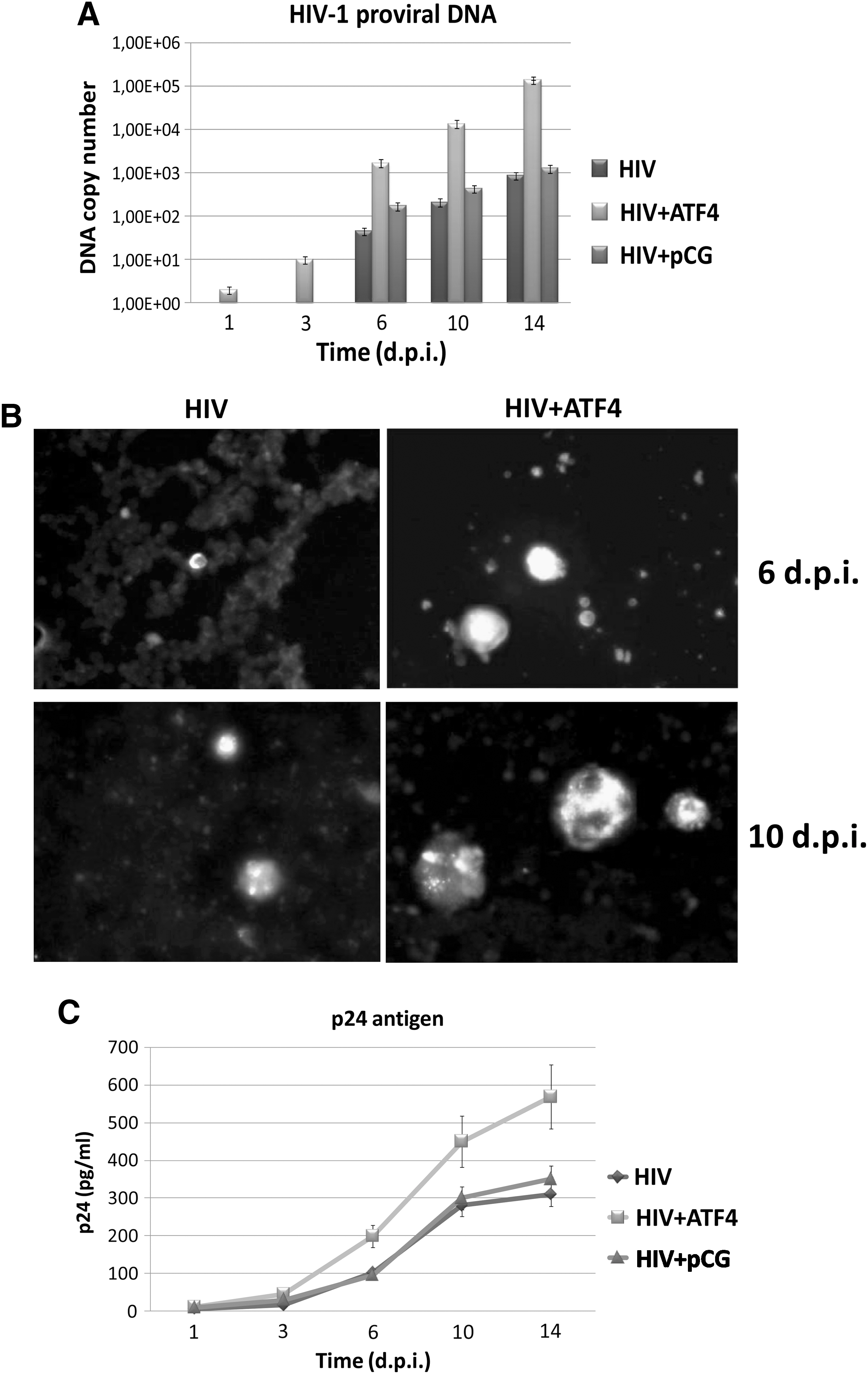
ATF4 increases HIV-1 replication. A total of 106 CD4+ Jurkat T cells were untreated or transfected with 1 μg of pCG-ATF4 plasmid 24 h before HIV-1 infection (moi 1:10,000). Control cells were transfected with the same amount of pCG empty vector. Samples were then collected at the indicated hours postinfection (hpi) to analyze retrovirus replication.
To establish if the higher intracellular contents of HIV-1 proviral DNA and p24 antigen correlated with enhanced production of retroviral particles, p24 release in culture supernatant was measured by ELISA. 26 This assay revealed that ATF4-transfected cells produced at least 2-fold more p24 antigen than control cells (p<0.01), confirming PCR and IFA results (Fig. 2C). The higher release of new particles was further confirmed by the quantitation of genomic HIV RNA in culture supernatant, obtained by RT-qPCR specific for the gag gene, performed on total RNA extracted from culture medium (not shown).
Since enhancement of HIV-1 replication is associated with transcriptional activation of LTR, we analyzed the ability of ATF4 to activate the HIV-1 promoter. Reporter assays were performed on Jurkat, 293, or HeLa cells. Cells were cotransfected with increasing amounts of pCG-ATF4 plasmid or pCG vector alone (control), and with 500 ng of an HIV LTR promoter-luciferase construct (pLTR-luc), obtained by cloning the HIV promoter in the pGL3 Basic vector (Promega Corporation, Madison, WI).
Cotransfections included the presence or absence of increasing amounts of a plasmid encoding the HIV tat gene (pRP-tat), 27 and a constant amount (500 ng) of pRL-SV40 plasmid (Promega Corporation, Madison, WI), used as an internal control. Efficiency of transfection was determined in parallel samples by transfection with pmax-GFP plasmid (Amaxa), and was approximately 60% in Jurkat and 80% in 293 and HeLa cells. Briefly, 106 cells seeded at optimal density were cotransfected by nucleofection (Nucleofector, Amaxa, Cologne, Germany), seeded in 24-well plates, and collected after 48 h for the analysis of luc activity using a Berthold Microplate Luminometer (Dual-Glo Luciferase Assay System, Promega). As shown in Fig. 3A, ATF4 in Jurkat T cells did not directly transactivate HIV LTR, but it synergized the action of tat. In fact, in the presence of suboptimal amounts of pRP-tat, ATF4 enhanced LTR activity up to 10-fold compared to cells that did not receive pCG-ATF4 plasmid (p<0.005). Similar results were obtained in epithelial 293 cells (not shown). Instead, the same experiments performed on HeLa cells showed that ATF4 transactivated HIV LTR even in the absence of tat, suggesting that its activity on HIV LTR is influenced by the cellular environment (Fig. 3B). Since in HeLa cells ATF4 can directly induce LTR activation, its action appears less dependent on synergism with Tat.

ATF4 enhances LTR activation.
Based on these observations, we investigated the effect of ATF4 expression on the reactivation of latent HIV. U1 promonocytic cells, chronically infected by inducible HIV, were seeded at 0.5×106 cells/ml in 12-well plates and nucleofected with pCG-ATF4 or pCG control empty plasmid. The efficiency of transfection was approximately 60%, as determined in parallel samples by transfection with pmax-GFP plasmid (Amaxa). As a positive control of HIV reactivation, cells were treated with TPA (20 ng/ml) or infected with a cell-free inoculum of HHV-8, 21 using an moi of 10:1. Culture supernatants were analyzed for HIV-1 reactivation by RT-qPCR for gag gene and by p24 ELISA. As expected, HHV-8 infection and TPA treatment induced a rapid and potent reactivation of latent HIV, visible both as HIV-1 genome number and p24 antigen increase in culture medium (Fig. 4A and B). More importantly, ATF4 also induced a significant increase of viral load in U1 cell culture supernatant (p<0.001 at 7 and 12 dpi), although with less efficiency compared to TPA or HHV-8.
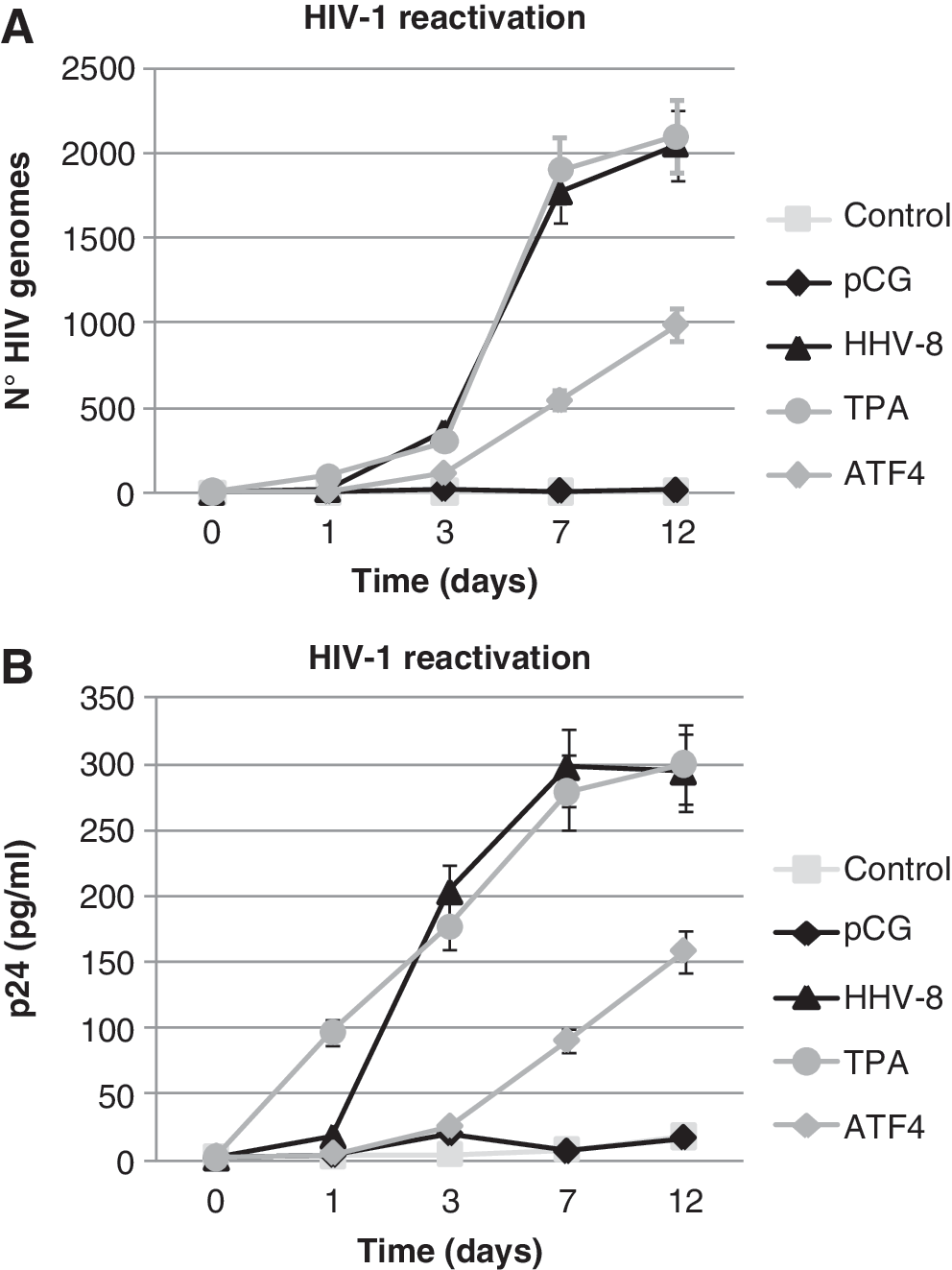
ATF4 induces HIV-1 reactivation in latently infected cells. The promonocytic U1 cells were untreated (Control), transfected with empty pCG (pCG) or pCG-ATF4 (ATF4) plasmids, infected with HHV-8 (HHV-8), or treated with TPA (TPA). HIV-1 reactivation was evaluated on cell culture supernatant at the indicated time points.
These results show that HIV-1 induces ATF4 transcription and translation, and causes ATF4 accumulation during acute infection. The induction of ATF4 is an early event in the course of HIV-1 infection, as shown by the detection of ATF4 mRNA and protein 8 hpi in HIV-infected CD4+ T cells, and is relevant in promoting HIV-1 replication. In addition, ATF4 overexpression reactivates HIV-1 in latently infected U1 cells, suggesting that in vivo events causing ATF4 increase, such as ER stress, might be relevant for reactivation of latent HIV-1.
Our data demonstrate that ATF4 alone does not activate directly HIV-1 LTR, but it significantly enhances Tat activity, especially with low Tat concentrations. A similar situation occurs in the case of HTLV-1, where activation of HTLV-1 LTR by CREB-2 occurs only in the presence of Tax, 23 and suggests that HIV-1 Tat might similarly interact with the bZIP domain of ATF4, stimulating HIV-1 LTR activity. By contrast, our data on HeLa cells show a direct ability of ATF4 to induce HIV-1 LTR activation, even in the absence of Tat. This different behavior might be related to the ability of ATF4 to form cross-family heterodimers with members of the AP-1 and C/EBP families, interacting with Jun, Fos, and C/EBP proteins, and originating complexes with different binding specificity. 28 The regulatory region of HIV LTR located downstream of the TAR sequence contains elements that can bind both AP-1 and CREB/ATF, allowing LTR activation by both PKC and PKA signals, depending on the cell type. 29 Thus, our results suggest that when other cellular/viral factors are overexpressed, a possible ATF4 heterodimer might be able to activate specific sites of HIV-1 LTR, even in the absence of Tat.
The results suggest that in vivo conditions leading to ATF4 expression might cause HIV activation. This is of particular interest in light of the reports showing that Tat protein contains a redox-sensitive cystein-rich region that is essential for its ability to transactivate, 30 that anoxic conditions increase HIV-1 RNA expression, 31 and that exogenously provided Tat can prime T cells for HIV-1 infection under hypoxic conditions. 32 These observations are relevant in the context of ER stress, since the coordinated ER and oxidative stress signaling find ATF4 a common downstream signal. 33 In addition, the activation of CD4+ T cells has been recently reported to involve the activation of an ER stress response, 34 and HIV-1 Vpr has been shown to induce ER stress in gut epithelial cells, suggesting that this mechanism might be relevant in AIDS enteropathy. 35
These results show for the first time that ATF4 induction might have an important role in HIV replication, and suggest that ATF4 might represent a convergent signaling molecule for different stressors important in regulating the HIV-1 cycle.
Footnotes
Acknowledgments
This work was supported by the Italian Ministry of University and Scientific Research (PRIN projects), FAR (University of Ferrara), and Istituto Superiore di Sanità (ISS; AIDS project). We thank Dr. T. Hai (Department of Molecular and Cellular Biochemistry, Neurobiotechnology Center, Ohio State University, Columbus, Ohio) for providing the pCG-ATF4 plasmid. We thank Annalisa Peverati for excellent technical assistance, and Linda M. Sartor for revising the English manuscript.
Author Disclosure Statement
No competing financial interests exist.