
Abstract
Intracellular levels of cyclic adenosine 3',5'-monophosphate (cAMP) are important regulators of immune cells, partially determining the balance between activation and suppression. In this review, we discuss the mechanisms by which HIV infection increases cAMP levels in T cells, as well as the effect of cAMP on HIV-specific responses and its effect on HIV replication and infection. Results suggest that increased cAMP levels during HIV infection may have a dual and opposite roles. On the one hand, they could have a protective effect by limiting viral replication in infected cells and decreasing viral entry. On the other hand, they could have a detrimental role by reducing HIV-specific antiviral immune responses, thus reducing the clearance of the virus and contributing to T cell dysfunction. Future studies are thus needed to further define the beneficial versus detrimental roles of cAMP, as they could help establish new therapeutic targets to combat HIV replication and/or identify novel ways to boost antiviral immune responses.
Introduction
V
Regulation of the cAMP Pathway
cAMP is a secondary messenger involved in multiple cellular processes. Activation of cAMP production is induced by signals given through many cellular receptors, mainly G-protein-coupled receptors, adrenergic and apurinergic receptors, as well as growth factor receptors. 1 Intracellular levels of cAMP are controlled by two groups of enzymes, those that induce it, the adenyl cyclases (AC), and those that degrade it, the phosphodiesterases (PDE). Ten different isoforms of AC (AC1–AC10) and 11 isoforms of PDE (PDE1–PDE11) have been reported. 2,3 T cell express different isoforms of AC (AC3, AC6, AC7, and AC9 4 ) and PDE (PDE1B, PDE3B, PDE4D, PDE8A, and PDE11 5,6 ). In general, AC are bound to the inner side of the cell membrane and, once activated, they transform ATP into cAMP. In contrast, PDE are mainly found in the cytoplasm and they hydrolyze cAMP to its inactive form, the adenosine 5′-monophosphate (AMP). 7
Of particular interest is the fact that regulatory T cells (Treg) can induce cAMP in their target cells by increasing adenosine levels in the microenvironment, through conversion of ATP into adenosine, a process mediated by ectonucleotidases (CD39 and CD73) present at the surface of Treg. 8 First, CD39 hydrolyzes ATP or ADP into 5-AMP, which is cleaved into adenosine by CD73. 8 Pericellular adenosine signals through the purinergic receptor A2AR, thus inducing AC activation in Treg target cells.
An additional mechanism of increased intracellular cAMP involves influx of cAMP from Treg, through gap junctions (GJ). GJ are channels that allow intercellular communication between adjacent cells; they are formed by two opposing hemichannels from each cell, called connexons. This protein complex consists of six proteins called connexins (Cx). 9 GJ are used for the bidirectional passage of ions, metabolites, and other molecules of less than 1 kDa. 9 Resting T cells exhibit low density of Cx31.1, Cx32, Cx43, Cx45, and Cx46, which all increase after cellular activation. 10 Previous studies have shown that Treg contain high levels of intracellular cAMP, which they can transfer through GJ to target cells, including T cells and dendritic cells (DC), and thus increase intracellular cAMP in these target cells. 10,11
cAMP activation initiates several major downstream signaling cascades. The canonical pathway is the activation of the cAMP-dependent protein kinase-A (PKA). More recently, cAMP was also shown to interact with the Exchange Protein Activated by cAMP (EPAC). A third major target is the Cyclic-Nucleotide Gated Ion Channel (CNG). Together or separately, these pathways regulate the transcriptional activity of many genes involved in cell cycle, cell survival, and cytokine secretion. 12 In addition to these two main pathways, cAMP directly regulates Ca2+ levels by opening ion channels. This controls T cell proliferation and cytokine production. 12
PKA acts on multiple signaling molecules inside the cells, thus inhibiting the transcription of many genes. It negatively regulates the transcription factor CREB, blocking the formation of the complex with the coactivator CBP, preventing the binding to the cAMP response elements (CRE). 13 These CRE binding elements are found in the promoter of many genes coding for the T cell receptor, CD3, and other molecules involved in T cell activation. 14 PKA also regulates the activity of NFAT, by blocking the interaction and the formation of protein complexes, and it blocks the formation of NF-κB and CBP/p300 complex. 15 In addition, PKA phosphorylate the proteins Raf-1, Ras, Mek, and HePTP in the MAPK pathway, as well as PLC-α1 and PLC-β in the phosphatidylinositol pathway. 15,16
The EPAC pathway regulates the expression of several genes associated with cell cycle, affecting T cell proliferation, as well as the production of cytokines such as IL-2 and IL-5. 17,18 Interestingly, anergic T cells express the active form of RAP-1, which is considered to function as a negative regulator of gene transcription induced by TCR engagement and IL-2. 19 In contrast, EPAC signaling does not appear to affect the maturation and function of DC. 20
cAMP Decreases T Cell Responses
In immune cells, the cAMP/PKA pathway directly modulates the proliferation and transcription of many cytokine genes, such as TNF-α, IFN-γ, IL-2, IL-4, IL-5, IL-6, IL-9, IL-10, and IL-12. 15,21 –23 Additionally, cAMP suppresses the proliferation of murine or human T cells stimulated with anti-CD3 antibody. 17,24,25 Treatment of T cells with cholera toxin activates AC by inducing an ADP-ribosylation in the alpha subunit of the G protein, leading to increased cAMP levels and decreased proliferation and IL-2 production in response to stimulation. In addition, cholera toxin promotes the acquisition of regulatory functions, 26 mediated by increased expression of the immune suppressive molecule cytotoxic T-lymphocyte antigen 4 (CTLA-4). 27
cAMP signaling can also indirectly regulate T cell function as induction of cAMP in DC following engagement of prostaglandin and adenosine receptors negatively regulates NF-KB activity, decreasing the production of proinflammatory cytokines such as IL-12, TNF-α, IL-1α, and IL-6, while increasing the production of the antiinflammatory cytokine IL-10. 28 –34 In addition, cAMP affects the expression of costimulatory molecules by DC and thus their immunogenicity. 29,33,35,36
Levels of cAMP in Treg also partially determine their suppressive activity. Antagonists of cAMP decrease Treg suppressive activity and increase antitumoral immune responses in patients with colon cancer. 37 However, treatment of Treg with rolipram, a PDE inhibitor that prevents cAMP degradation, potentiates their suppression of Th2 cells in vivo, controlling tissue inflammation and allergic respiratory diseases. 38 Inhibition of the cAMP pathway could thus constitute a therapeutic avenue in diseases in which an excessive immune regulation exerted by Treg plays a pathogenic role.
HIV Increases Intracellular cAMP
Previous studies have shown that in vitro HIV infection of T cell lines and primary T cells leads to enhanced intracellular cAMP levels. 39,40,41 Moreover, ex vivo studies have shown that T cells from HIV-infected patients contain twice as much cAMP than those of HIV-uninfected individuals. 41,42 Of note, these studies were done before it was recognized that Treg frequency increases during chronic HIV infection, 43 –46 so these later results may be, at least partially, due to the changes in subset composition. Productive infection was not required, as treatment of T cells by the HIV envelope glycoprotein 120 (gp120) was sufficient to augment cAMP levels and activate the cAMP/PKA signaling pathway. 42,47,48 How HIV affects EPAC is not well understood. Similarly, the mechanism(s) by which gp120 increases cAMP are still unclear. One study described the binding of HIV gp120 to chemokine receptors as the driving mechanism, as engagement of CD4 by a specific antibody did not have the same effect as CXCR4 engagement by its natural ligand SDF-1. 47 In contrast, another study suggests that cAMP induction by gp120 is mainly CD4 dependent, as Lck blockage abolished its induction. 48
HIV Can Directly Activate the Extracellular Pathway
In addition to directly augmenting intracellular levels of cAMP, HIV could also increase cAMP levels by modulating the production of adenosine through the regulation of CD39 expression. CD4+ T cells from untreated HIV-infected patients exhibit an increase in ATPase activity, a result that was associated with a higher percentage of CD39+ T cells. 49 Increased CD39 expression by Treg was reported in HIV-infected progressors compared with healthy controls and elite controllers. 50,51 Moreover, during the acute phase of SIV infection in rhesus macaques, CD39 was highly expressed by the CD8+FOXP3+CD25+ T cells present in the gastrointestinal mucosa and lymphoid organs, which are sites of active viral replication. 52 This increased CD39 expression on T cells may limit viral infection by decreasing levels of extracellular ATP. Indeed, ATP has recently been shown to facilitate viral entry, as it promotes the fusion of viral membrane and host cell membrane in a pathway involving binding of ATP to purinergic receptors. 53
cAMP Decreases Anti-HIV T Cell Responses
Several studies have shown that activation of the cAMP pathway could inhibit HIV-specific immune responses. In particular, gp120 binding to CXCR4 decreased the proliferation of CD4+ and CD8+ T cells in response to polyclonal stimuli, through PKA-dependent activation of CREB. 40,47 In contrast, decreasing intracellular cAMP generation by chemical inhibition of AC restored the proliferation and cytotoxicity of T cells. 40,42 Another mechanism of cAMP-mediated suppression is its effect on Treg: HIV gp120 potentiated Treg-mediated suppression by increasing CTLA-4 levels on these cells. 48 Moreover, blocking CD39 activity boosted the production of cytokines by HIV-specific T cells. 51 Taken together, these results may explain the relative protection against development of AIDS associated with a CD39 gene polymorphism that leads to low CD39 expression. 51
cAMP Affects HIV Replication
All together, the studies mentioned above suggest that cAMP plays a detrimental role during HIV infection because it can suppress the antiviral immune responses. However, other data suggest that it plays a more complex role as it can also directly inhibit viral replication (Fig. 1). Increased levels of cAMP either following in vitro AC activation with synthetic compounds such as forskolin, or after blockage of cAMP degradation by rolipram, diminished viral transcription and levels of HIV-p24Gag protein in activated T cells. 54,55 In addition, ATP treatment of HIV-infected immature DC induced lysosomal degradation of the virus and blocked virus transfer from DC to CD4+ T cells. 56 In infected primary T cells, monocytes, and cell lines, cAMP activates the CREB protein, which competes with phosphorylated NF-κB for limiting amounts of CBP/p300, suppressing HIV-LTR transcription activity in infected cells. 57 –59 In naive T cells, cAMP significantly decreased nuclear import, translocation, and replication of viral DNA, compared to memory T cells, suggesting that the cAMP/PKA pathway can affect HIV infection at both pre- and postintegration steps. 55
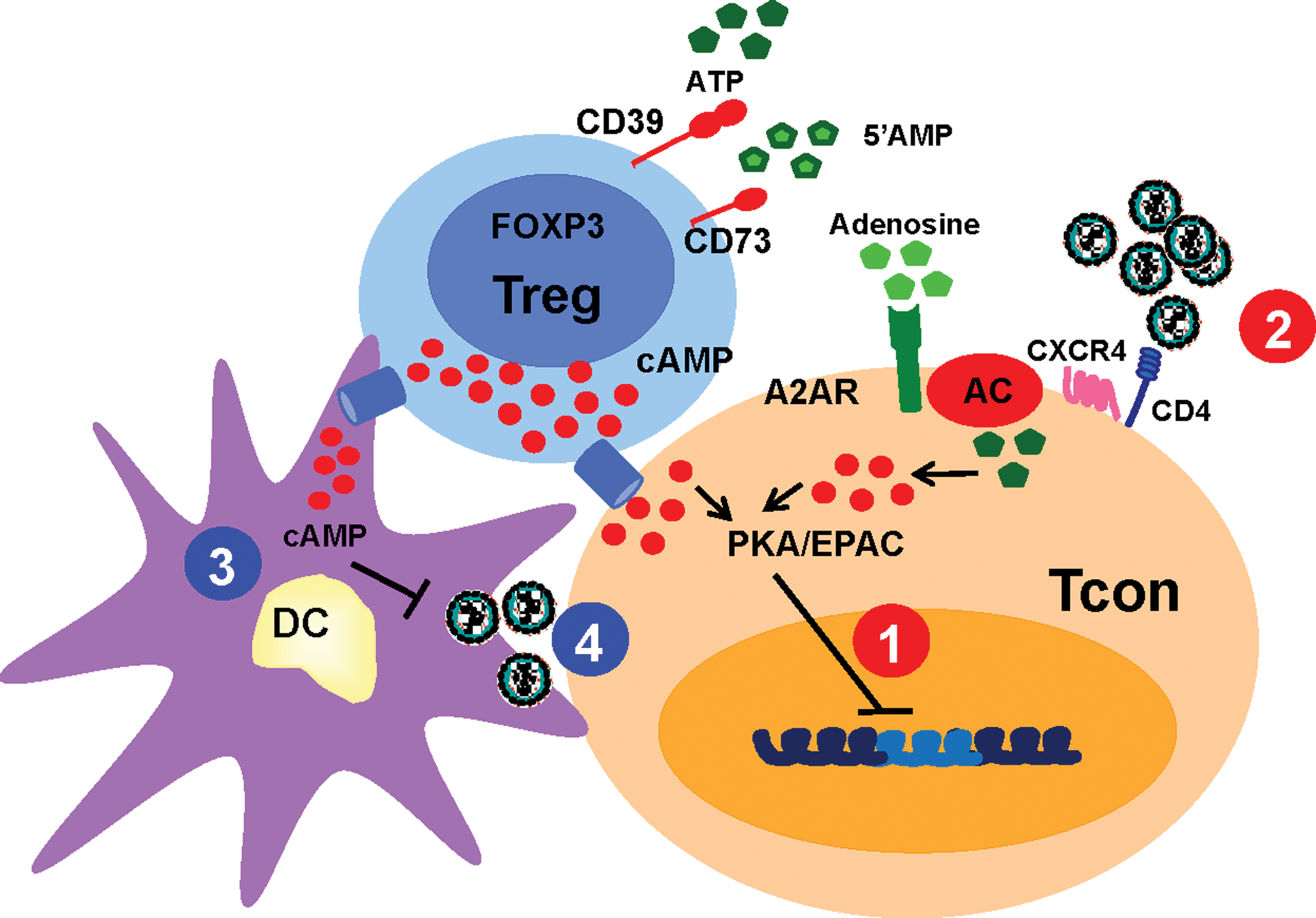
Effect of increased cAMP levels in HIV replication. The scheme illustrates the various mechanisms by which increased levels of cAMP may affect HIV replication and spread. The red circles represent the effect of increased cAMP in conventional T cells (Tcon), either by a direct effect of the virus or by transfer of cAMP from the Treg. These mechanisms include
cAMP could also limit HIV infection by decreasing the expression of viral receptors on the surface of target cells. In particular, stimulation of the PGE2 receptor, which induces cAMP in monocytes and macrophages, decreases their expression of CCR5. 60 Similar regulation occurs in CD4+ T cells, in which activation of the cAMP pathway following stimulation of adenosine receptors reduces the expression of the coreceptors CXCR4 and CCR5. 61
Another level of control of HIV replication that involves cAMP is the one exerted by Treg. Recently, using an in vitro model of HIV infection, we showed that Treg limit HIV infection in conventional T cells using a cAMP-mediated pathway involving both transfer of cAMP through GJ and CD39 activity. This reduction of HIV infection in conventional T cells implicated the PKA pathway, because inhibition of PKA activation in target cells abolished Treg suppression of HIV infection. 62 Furthermore, cAMP from Treg likely inhibits DC activation as Treg and DC form GJ. 11 As the level of activation of DC is a critical factor in their transfer of virus to CD4+ T cells, increased cAMP in DC may also limit this transfer. This function of Treg has not yet been investigated, but its potential to limit early viral spread warrants further investigation. Of note, the effect of cAMP-mediated EPAC activation on HIV immune responses or HIV replication is not yet established. However, as EPAC controls the expression of many genes involved in T cell activation and cell cycle, this pathway also deserves further investigation. 63
Conclusions
Experimental evidence suggest that cAMP has dual and opposite roles during HIV infection. On the one hand, it could have a protective effect in infected cells by limiting viral replication, decreasing viral entry, or decreasing the capacity of DC to transfer the virus. On the other hand, it could have a detrimental role by reducing HIV-specific antiviral immune responses, thus reducing the clearance of the virus and contributing to T cell anergy. The balance between these functions is not known, but one could envision a model whereby the overall effect of cAMP is dependent of the stage of the infection. During the early HIV infection when the effector immune responses are not yet established, cAMP could have a mainly beneficial role, controlling T cell and DC activation, thus decreasing viral replication and viral transfer. In contrast, during the late and chronic phase of the infection when anti-HIV effector responses have been primed, high cAMP levels could decrease their efficiency, and have an overall negative effect despite its continuous capacity to decrease viral replication. It is therefore important to design more studies to evaluate the effect of cAMP activation at different stages of HIV infection. This knowledge may make it possible to establish new therapeutic targets of antiretroviral therapy or identify potential target molecules with immunoregulatory potential, which could help restore immune dysfunction.
Footnotes
Acknowledgments
This work was supported by Public Health Service Grants AI068524 (to C.C.) and by Colciencias 111540820490-1 (to C.M.R.). We thank Drs. Dave Hildeman and Gene Shearer for critical review of this manuscript.
Author Disclosure Statement
No competing financial interests exist.