
Abstract
We studied 123 samples from adult chronic HIV patients initiating HAART from various centers around a newly established clinical trial site in Pretoria. Each sample was sequenced in at least one structural gene (pol, gag, and env) or functional gene (vif, vpr, and vpu). A subset of 25 samples was subjected to near full-genome analysis. All samples were HIV-1 subtype C. Highly conserved regions within the gene sequences were observed. Overall, the gag and vif sequences showed closer similarity followed by the env, vpr, pol, and vpu. The env gene was the most difficult to sequence, resulting in only 31 sequences from 40 samples; of these, 25 were predicted to be R5 coreceptor tropic, while 6 were X4 tropic. The study asserted the predominance of HIV-1 subtype C within the catchment population.
The United Nations program on HIV and AIDS (UNAIDS) estimates that there were 33.3 million (31.4–35.3 million) people living with human immunodeficiency virus (HIV) worldwide at the end of 2009. Sub-Saharan Africa still bears the greater share of the global HIV burden. Southern Africa is the most affected with an estimated 11.3 million (10.6–11.9 million) people living with HIV in 2009. South Africa's epidemic remains the largest in the world, with an estimated 5.6 million (5.4–5.8 million) people living with HIV in 2009. 1
HIV diversity is driven by the characteristic nature of the virus to evolve rapidly, giving rise to substantial genetic diversity among different isolates. 2 This is attributed in large part to the high mismatch error rate (approximately 3.4×10−5 mutations per base pair per replication) during reverse transcription due to the lack of exonuclease proofreading activity of reverse transcriptase (RT). 3 Genetically, HIV is classified into types, groups, subtypes, subsubtypes, circulating recombinant forms (CRFs), and unique recombinants with the global pandemic caused by the M group viruses. The region with the highest HIV prevalence in Africa is southern Africa, which is predominantly a subtype C region, with prevalence as high as 98.3% in Zambia, 4 almost 100% in Zimbabwe 5 and Botswana, 6 and 91.5% in South Africa. 7
The only lasting solution to any viral pandemic is a preventive vaccine. The development of an HIV vaccine has been the main focus of many scientific groups and has so far proved to be the most difficult. Globally, there has been a rise in the number of HIV vaccine candidates going into clinical trials. South Africa has seen more than six clinical trials including two HIV-1 subtype C-based vaccines developed in South Africa (SAAVI MVA-C and SAAVI DNA-C2) 8 and the large phase IIb HVTN503/Phambili trial that tested a subtype B-based vaccine.
Currently, over five sites are involved in HIV vaccine trials in South Africa; one of them is a newly established Medunsa Clinical Research Unit (MeCRU), located at Medunsa Campus of the University of Limpopo in Pretoria. However, the impact of HIV-1 subtype C (and other subtypes)-based vaccine candidates on the genetic diversity of prevalent HIV subtypes at a clinical trial site is not known. This study aimed to generate data on the genetic landscape of prevalent HIV strains before the widespread testing of vaccine candidates in the Pretoria region.
A total of 123 archived (−70°C) HIV-positive plasma samples collected between 2006 and 2007 were used for the study (Table 1). The samples were from adult chronic HIV patients initiating highly active antiretroviral therapy (HAART) from various treatment centers around Pretoria, and were selected from the Department of Virology diagnostic laboratory of the National Health Laboratory Service (NHLS) at Dr. George Mukhari Tertiary Laboratory after routine testing for HIV viral load. Only samples with HIV viral loads of 100,000 copies/ml or greater were selected.
Overview of the Study Population and Gene Targets Amplified and Sequenced (N=123)
Subset of 25 samples contributed toward full-genome analysis.
All 25 samples were part of the full-genome analysis.
The NHLS Dr. George Mukhari Tertiary Laboratory is located 31 km northwest of Pretoria at Medunsa Campus, University of Limpopo. For the purpose of this study, only samples from the neighboring townships of Pretoria including Ga-Rankuwa, Soshanguve, Mamelodi, Laudium, Kalafong, Jubilee, and Mabopane were included. These townships would be preferred recruitment areas for the newly established MeCRU. Approval for the study was granted by the Medunsa Research and Ethics Committee (MREC) (project number MREC/P/136/2008: PG).
RNA extraction was performed from 200 μl of plasma using the QIAamp Viral RNA Mini Kit (Qiagen, Valencia, CA). RNA was eluted in 60 μl and stored at −70°C until use. The accessory genes (vif, vpr, and vpu) were amplified as one target, while the complete major genes (gag, pol, and env) were each amplified separately. cDNA synthesis was performed using the revertAid kit (Fermentas, MD) following the manufacturer's instructions.
This was followed by first and second round polymerase chain reaction (PCR) reactions performed in a total of 25 μl containing 0.4 μl of 25 mM dNTPs (Fermentas), 0.5 μl of 10 mM primer 1, 0.5 μl of 10 mM primer 2 (Table 2), 2.5 μl of 10×buffer, 0.2 μl of Expand long template DNA polymerase (Roche Diagnostics, Penzberg, Germany), and distilled water, which was added depending on the amount of template to make a 25 μl reaction mix.
PCR and Sequencing Primers
PCR, polymerase chain reaction.
The amplification was performed in an Applied Biosystems 2720 thermocycler with the following cycling conditions: initial denaturation at 94°C for 1 min, followed by 10 cycles each of denaturation at 94°C for 20 s, annealing temperature (Table 2) for 30 s and elongation at 72°C for 2.30 min, with an additional 25 cycles of the same cycling conditions with 10 s increment on elongation. The final elongation was set at 72°C for 10 min. Positive and negative controls were included in all amplification reactions, and necessary precautions were undertaken to avoid contamination.
Of the 123 samples, 92 were sequenced in the gag (p17/p24-p6), 51 in the pol (Prot-RT), 40 in the env (gp 120), and 25 in the accessory genes (vif, vpr, and vpu). A subset of 25 of 123 samples was subjected to near full-genome analysis on the major structural (pol, gag, and env) and accessory genes (vif, vpr, and vpu) (Table 1).
PCR products were sequenced with the SpectruMedix SCE 2410 Genetic Analysis System (SpectruMedix LLC, PA) employing gene-specific primers (Table 2). The nucleotide sequences were viewed using Chromas version 1.45 (School of Health Science, Griffith University, Australia).
Full length HIV-1 subtypes A, B, C, and D reference sequences were downloaded from GenBank. For each HIV subtype, 10 sequences from different geographic regions were selected. The generated sequences were aligned separately with HIV-1 subtypes A, B, C, and D reference strains from the Los Alamos sequence database using the BioEdit program. The sequences were analyzed on their potential antigenic regions according to the BioAfrica's HIV-1 proteomics resources tool. Comparison and alignment were done using MULTALIN multiple sequence alignment with hierarchical clustering.
Phylogenetic analysis was performed for each gene. The REGA program (
HIV Coreceptor Prediction for 31 env Sequences with WebPSSM Analysis Showing That 6/31 Samples Were Predicted to Be X4 Tropic While the Rest Were R5 Tropic
All the 123 samples subtyped in at least one of the major genes were HIV-1 subtype C. The sequencing success rate was very good in all the genes except the env, where only 31 of 40 samples could be successfully sequenced. The samples that failed sequencing in one region were successfully sequenced in the alternative gene(s). Phylogenetic analysis of the 25 near full-genome sample sequences confirmed that their sequences were HIV-1 subtype C (Fig. 1). Overall, the gag and vif genes sequences showed closer DNA sequence similarity with an average homology distance of 92.5% (89% to 96%) and 92% (88% to 96%) respectively, as compared to vpr gene 89% (84% to 94%), the env gene 88% (83% to 93%), the pol gene 87% (79% to 95%) and the vpu gene 82.5% (73% to 92%).
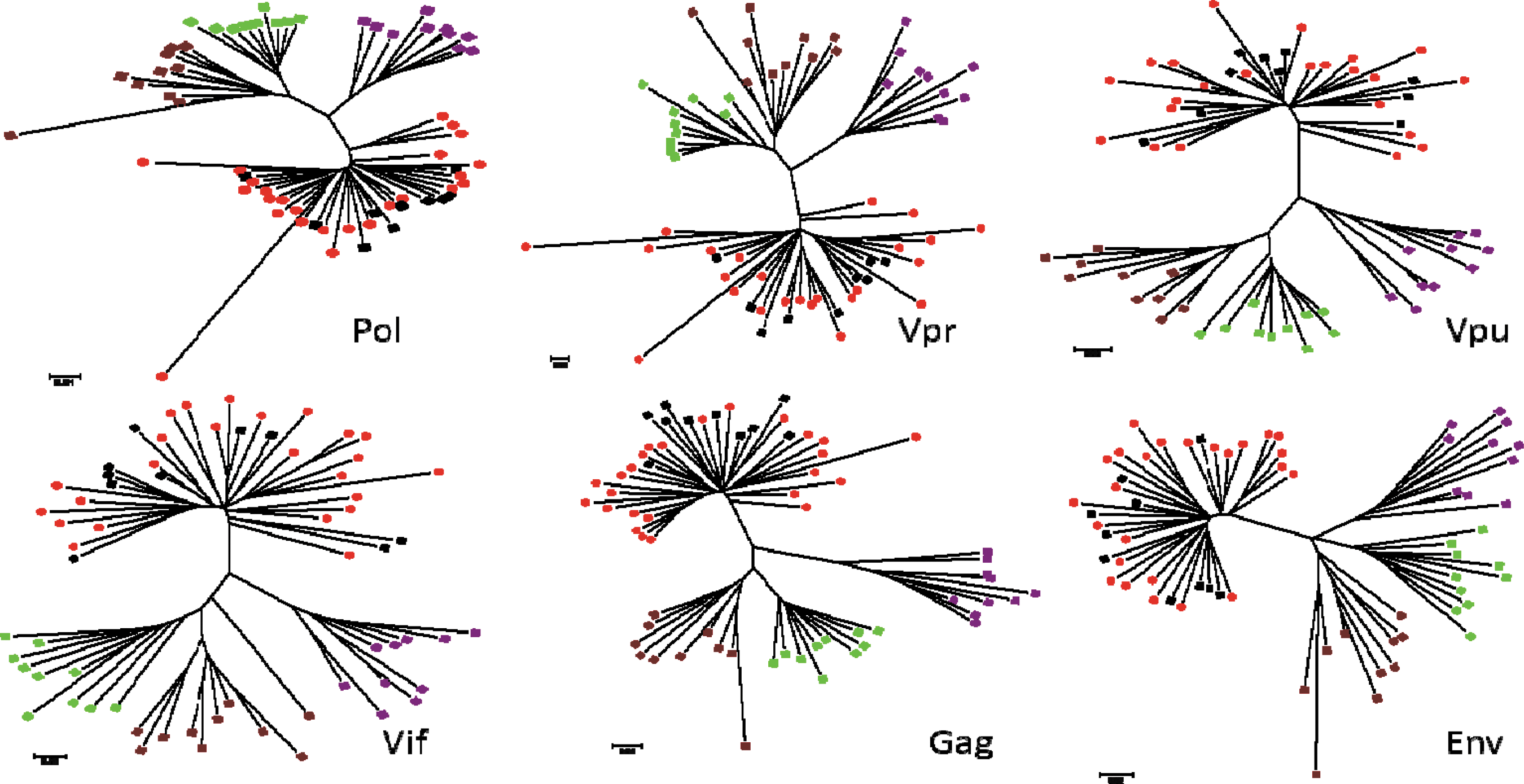
Phylogenetic analyses of a set of 25 samples on the pol, vpr, vpu, vif, gag, and env genes. Study samples are in red. HIV-1 subtype references are from GenBank. Subtype A=purple, subtype B=green, subtype C=black, and subtype D=maroon.
A total of 91 sequences were generated for the Gag region (Table 1). Functional and mutational analyses were performed for the p24-p6 region (39 sequences). Gag p24 was a relatively conserved gene with variability mainly found within p2 (aa 370) to p7 (aa 393). Of the 10 potential immunogenic sites within the sequences analyzed for differences in the aa composition, the sites corresponding to aa 123 to 129, aa 131 to 145, aa 147 to 154, and aa 227 to 286 had the least differences in the aa composition. Mutations in the HLAB57/5801 TW10 Gag epitope (gag aa 240 to 249) have been associated with fitness cost. This epitope, however, was conserved within these sequences with T242N mutations occurring in only 7/39 samples.
The cytotoxic T lymphocyte (CTL) epitopes were evaluated for conservation in 25 sequences selected for near full-genome analysis (Figs. 2 and 3). The 73-KLVDFRELNK-82 epitope was conserved in subtypes A, B, and C, with few variants in the test sequences and subtype D reference sequences. Another epitope, 260-LVGKLNWASQI-271, had one variant in one test sequence and one subtype B reference sequence. Subtype D reference sequences for the previous epitope had more variants than other subtypes. 179-VIYQYMDDL-187 showed more variants in subtype A and in 93-GIPHPAGLK-101 more variants were observed in the test sequences (Fig. 2).
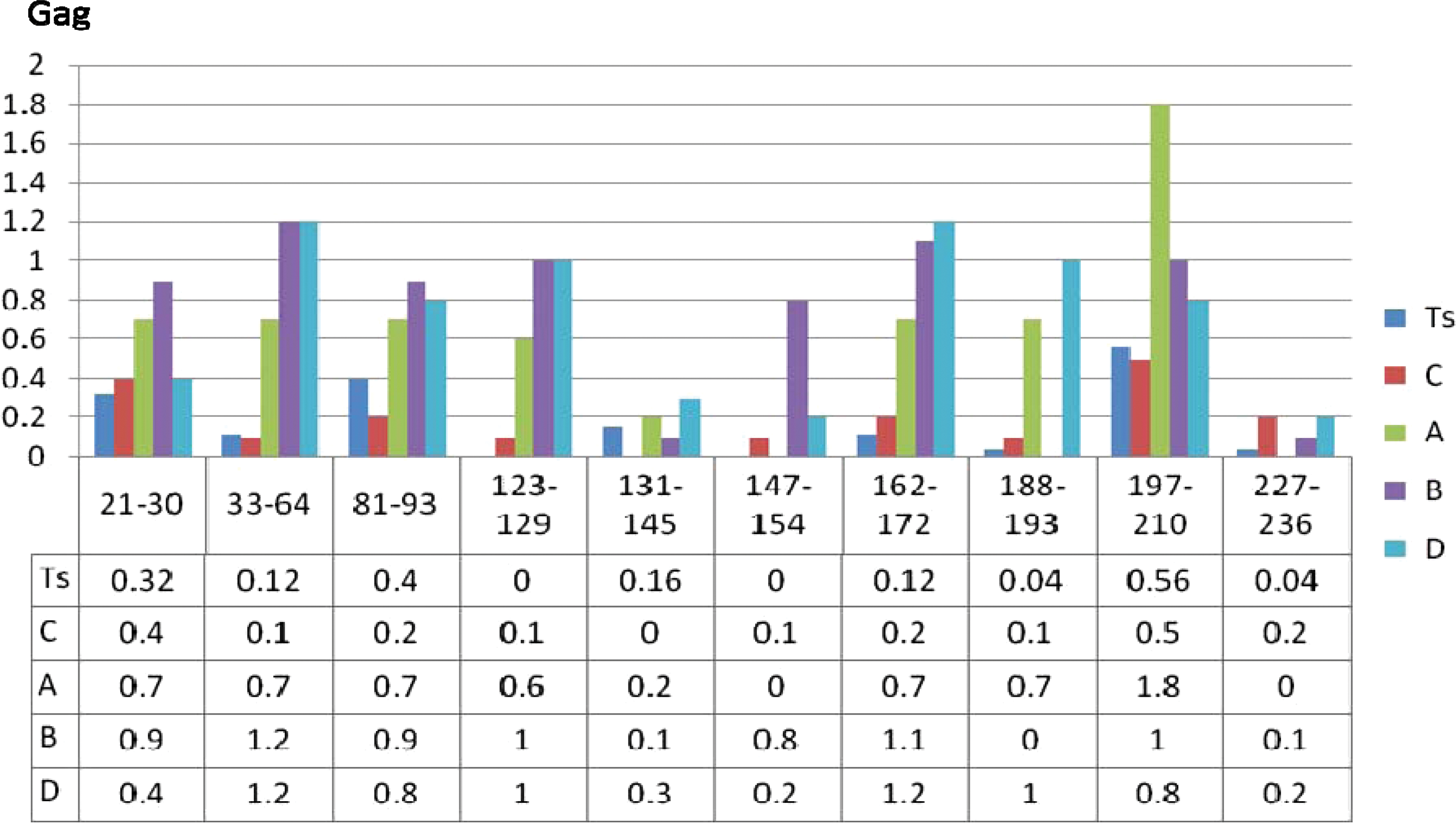
Amino acid (aa) variations within potential antigenic sites on the gag gene of test sequences (TS) and other HIV-1 subtypes. The x axis represents mutation frequency and the y axis represents antigenic sites numbered according to the HXBr coding sequence from the start of the gag gene. Color images available online at
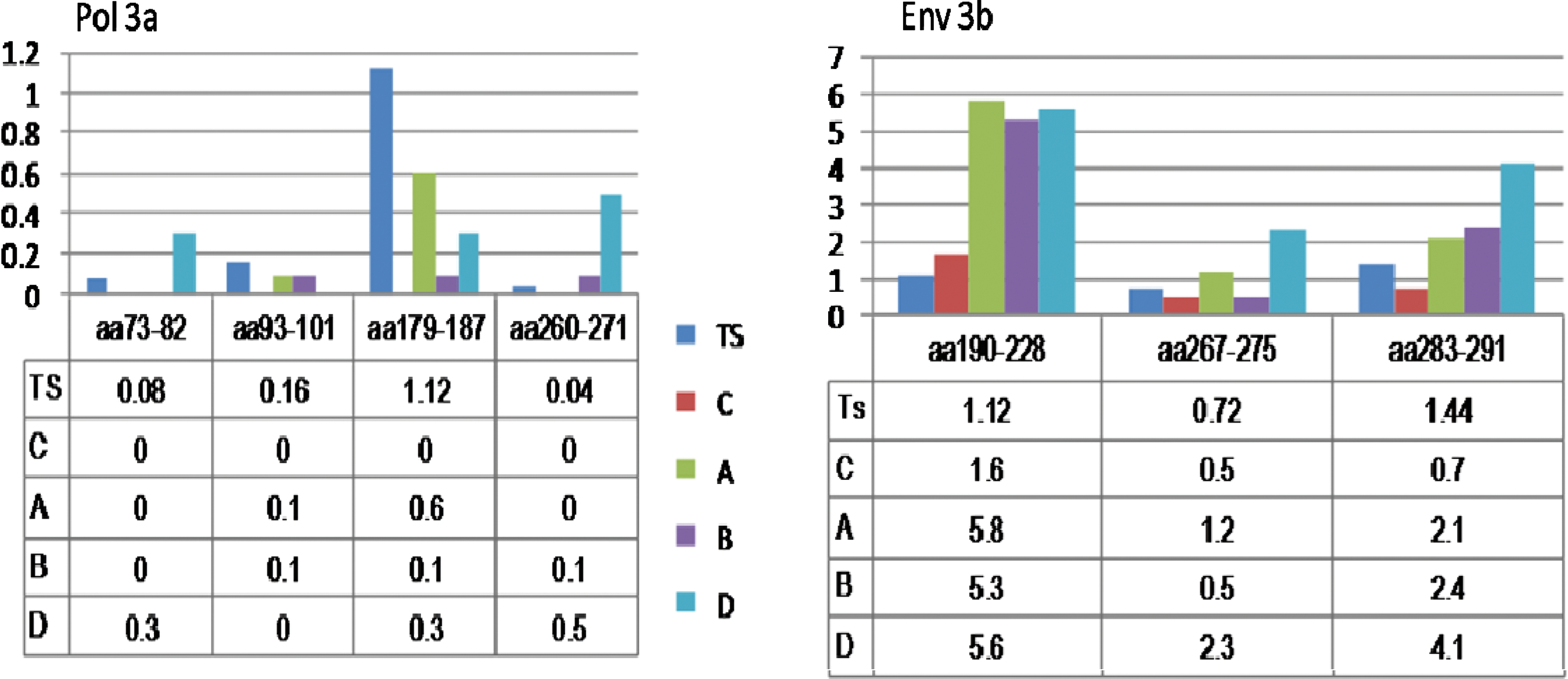
Amino acid (aa) variations within potential antigenic sites on the pol and env genes of test sequences (TS) and other HIV-1 subtypes. The x axis represents mutation frequency and the y axis represents antigenic sites numbered according to the HXBr coding sequence from the start of the Pol and Env genes respectively. Color images available online at
The env sequences covered the major determinant of the coreceptor specificity V3 loop within the gp120 gene. The V3 loop sequences were used to predict coreceptor tropism (Fig. 4). Six of 31 env sequences (19.4%) were potentially CXCR4-utilizing viruses, while the other 25 sequences (80.6%) were predicted to use CCR5 (Table 3). In addition, the 25 samples subjected to near full-genome analysis were investigated for three potential immunogenic sites. Overall, results indicated that these regions are relatively conserved across strains (Fig. 3).
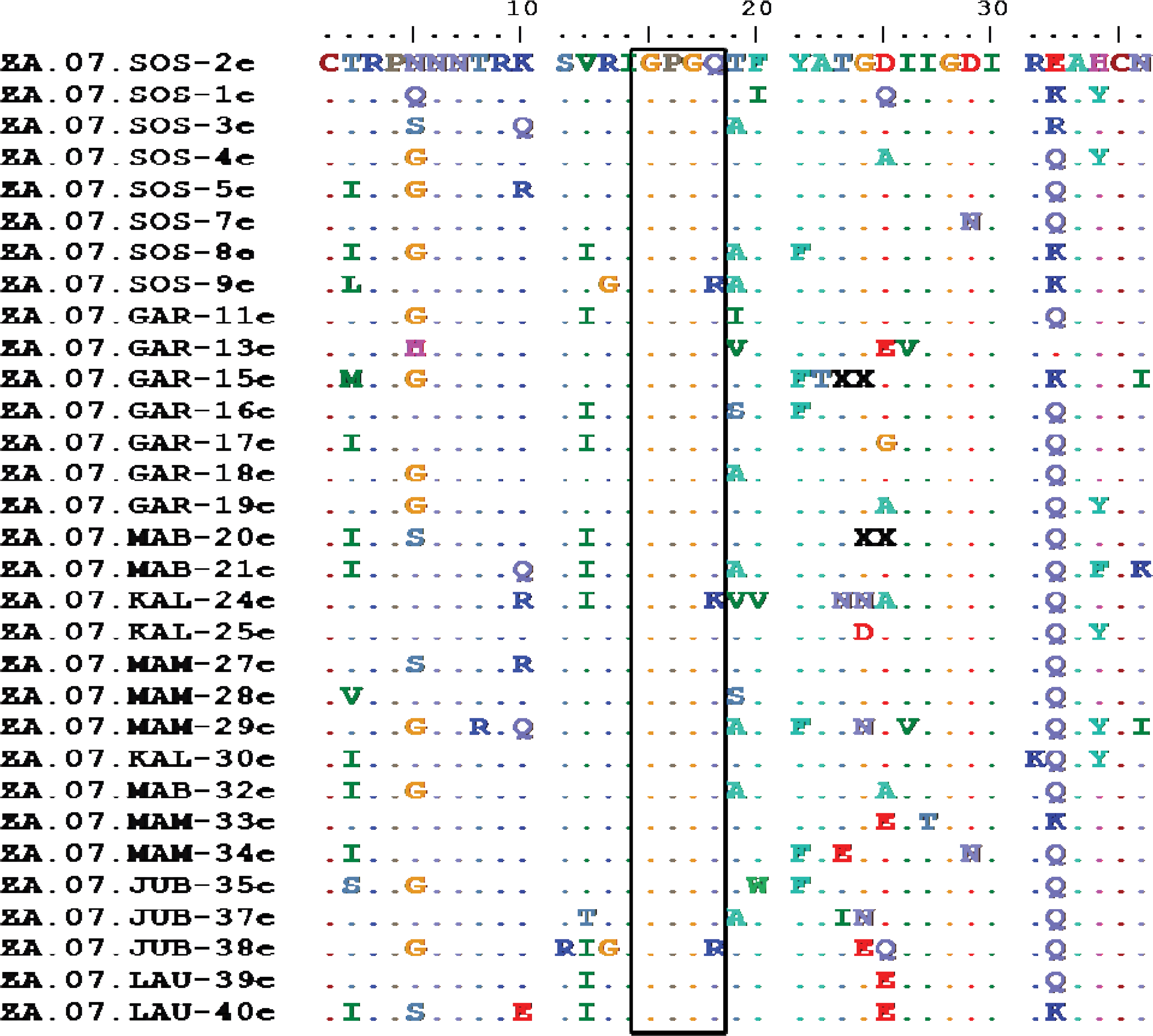
V3 loop sequences of study samples. Sample 2e was used as a reference. The HIV-1 subtype C characteristic GPGQ motif remained conserved in all but samples 9e, 24e, and 38e. Color images available online at
Sequence analysis of the vif gene revealed a high conservation including the cul5 binding HCCH (H108, C114, C133, H139) motif that functions to coordinate zinc and the BC-box 144-SLQYLA-149 motif and vif dimerization 161-PPLP-164; sequence residues W5, W11, W21, and W38 tryptophans, which are involved in recognition and suppression of APOBEC3G, were shown to be conserved in all the test sequences except the W38G variant in one test sequence JN176229. The previously identified conserved motif 90-RLRR-93 and vif dimerization site S95 showed more variations in the test sequences, which support findings by Jacobs and colleagues. 9
Mutations Q3R and R77Q on the vpr gene are associated with long-term nonprogression. 10 Mutation Q3R is also associated with high viremia with no significant loss of CD4 lymphocytes. 11 Of the 25 test sequences, two had mutation Q3R and 15 had mutation R77Q. One test sequence (JN176260) had both the Q3R and R77Q mutations.
The important residues in the vpr gene showed some degree of conservation. P35 and H71 were shown to be conserved in most of the test and reference sequences. The latter residues are involved in cyclophilin A binding and vpr dimer stacking, respectively. It was clear that important residues in the vpr gene were shown to be conserved though one or two variants were seen in some test and reference sequences.
The important residue and motif in the vpu gene, W28 and 57-DSGNES-62, were conserved. Only the 71-TMVD-74 and 78-LRLL-81 motifs showed many variants in both test and reference sequences.
This study sought to characterize prevalent HIV-1 strains from a recently developed clinical trial site in South Africa. All the samples tested were HIV-1 subtype C. The most plausible explanation for this observation is that South Africa, including the neighboring southern African countries, is predominantly HIV-1 subtype C.
The gag is intolerant of mutations since it codes for crucial structural proteins that are needed for production of complete infectious viral particles. For example, escape mutation T242N in the TW10 epitopes has been associated with reduced viral replicative capacity. 12 In our data set, mutation T242N appeared in 7 out of 39 samples. The knowledge of the high conservation of Gag-p24 and the predictable nature of escape variations resulting from these tight functional constraints indicate that p24 may be a critical immunogen in vaccine design and may suggest novel vaccination strategies to limit viral escape options from such epitopes.
Potential antigenic regions within the sequences were identified as described in the BioAfrica website and analyzed for variations between the sequences. The same was done for reference sequences from HIV-1 subtypes A, B, C, and D. Three regions were found to be most conserved. Regions spanned by aa 227 to 286, aa 131 to 145, and aa 147 to 154 had the least variation of the 10 regions analyzed. They had a total of 6, 10, and 11 aa variations, respectively. An effective vaccine will need to possess high cross-reactivity with other subtypes. These three regions seem to have the desired conservation across the subtypes prevalent in the sub-Saharan region.
The coreceptor binding site in gp120 is centered in the so called a “bridging sheet domain” formed from conserved discontinuous regions of gp120. The bridging sheet together with the third variable loop (V3) mediates coreceptor binding. Mutations within either of these domains can reduce the efficiency of coreceptor binding. In all the 25 samples tested, 23 were predicted to be R5 tropic. Two samples, ZA.07.MAM.29e and ZA.07.JUB.38e, were predicted to be X4 tropic. HIV-1 subtype C is known to maintain an R5 tropism for much of the course of infection 13 ; however, as these samples were obtained from people with very high viral loads it is possible that these individuals had AIDS. The V3 loop has also been established to be a major determinant of coreceptor specificity. As previously reported by Bessong et al., 14 analysis of the V3 loop across all samples revealed a conserved tetrapeptide motif GPGQ (aa 313 to 316) that is reported to be characteristic of HIV-1 subtype C strains. With the exception of samples ZA.07.SOS-9e, ZA.07.KAL-24e, and ZA.07.JUB-38e, which had mutations Q316R, Q316K, and Q316R, respectively, all samples maintained the GPGQ characteristic motif (Fig. 4).
Direct observation of the vif amino acid alignment sequences revealed a great degree of conservation. Motifs and residues within vif that were shown to serve important functions in HIV pathogenesis were highly conserved. These amino acids are involved in the steady-state expression of vif. The newly identified 69-YXXL-72 conserved motif was also found to be conserved in the sequences analyzed, and it also counteracts the action of APOBEC3G. 15 There were 20 variants in motif 90-RLRR-93 that were observed in the test sequences. The important residues in the vpr gene showed some degree of conservation. P35 and H71 were shown to be conserved in most of the test and reference sequences. The latter residues are involved in cyclophilin A binding and vpr dimer stacking, respectively. 9 It was clear that important residues in vpr were shown to be conserved, although one or two variants were seen in some sequences.
The important residue and motif in the vpu gene, W28, which is involved in channel gating, and 57-DSGNES-62, which is a casein kinase II phosphorylation site and contains two critical serines needed for CD4 degradation, were conserved. And the 71-TMVD-74 and 78-LRLL-81 motifs showed considerable variants in both test and reference sequences. Four immunogenic regions in the pol gene were analyzed for variations and region 73–82 was conserved in subtype A, B, and C. Only two variants were observed in the test sequences.
The process of formulating a vaccine takes a number of years to complete. Although the sequences discussed in this article may not represent presently circulating viruses, they offer a valuable insight into the genetic makeup of the predominant HIV strains in this area. Most importantly, some of the observed conserved genomic regions may be of interest to future vaccine development.
In conclusion, the study demonstrated that the majority of the predominant strains within the catchment population at the Pretoria clinical trial site are HIV-1 subtype C. The observation that most of the prevalent strains are CCR5 tropic suggests a potential for the successful application of R5 inhibitors. Comparison of immunogenic sites revealed potential sites that could be targeted for development of vaccine candidates with cross-reactivity to other HIV subtypes.
Sequence Data
The sequences were submitted to GenBank and are available under accession numbers JN167423 to JN167492, JN176214 to JN176310, and JF820610 to JF820662
The HIV-1 reference sequence accession numbers are as follows:
Footnotes
Acknowledgments
We thank the NHLS Virology Diagnostic Laboratory for technical assistance. The study was financially supported by grants from the Medical Research Council/South African AIDS Vaccine Initiative, Department of Science and Technology, and National Research Foundation in South Africa.
Author Disclosure Statement
No competing financial interests exist.