
Abstract
Cellular proteins are essential for human immunodeficiency virus type 1 (HIV-1) replication and may serve as viable new targets for treating infection. Using gene trap insertional mutagenesis, a high-throughput approach based on random inactivation of cellular genes, candidate genes were found that limit virus replication when mutated. Disrupted genes (N=87) conferring resistance to lytic infection with several viruses were queried for an affect on HIV-1 replication by utilizing small interfering RNA (siRNA) screens in TZM-bl cells. Several genes regulating diverse pathways were found to be required for HIV-1 replication, including DHX8, DNAJA1, GTF2E1, GTF2E2, HAP1, KALRN, UBA3, UBE2E3, and VMP1. Candidate genes were independently tested in primary human macrophages, toxicity assays, and/or Tat-dependent β-galactosidase reporter assays. Bioinformatics analyses indicated that several host factors present in this study participate in canonical pathways and functional processes implicated in prior genome-wide studies. However, the genes presented in this study did not share identity with those found previously. Novel antiviral targets identified in this study should open new avenues for mechanistic investigation.
Introduction
HIV-1
The majority of cellular targets implicated in HIV-1 replication have been identified in genomic siRNA screens. Due to the large numbers of genes tested, most genomic siRNA screens have employed reporter systems, replicon systems, or pseudotyped viruses that identify cellular factors important for a limited subset of stages in the viral life cycle. The predesigned siRNA libraries used for global screens have not included siRNAs targeting many unknown/hypothetical genes or nonprotein coding RNAs. We have used gene-trap insertional mutagenesis 17 as a complementary high-throughput screening (HTS) forward genetics approach to discover mammalian genes mediating infection, by identifying genes whose disruption confers resistance to otherwise lytic viruses. 18 –21 Using this technology, cellular genes are disrupted (trapped) with a recombinant Maloney murine leukemia virus (MMLV) virus encoding a promoterless neomycin resistance gene that randomly integrates into the chromosome. Integration of the MMLV vector between cellular promoters and early exons allows neomycin selection and derivation of gene-trap libraries with subverted gene expression. Infecting gene-trap libraries with a lytic virus typically results in >99.99% cytopathic effects (CPE), allowing high stringency selection of clonal cell lines surviving infection. Trapped genes in resistant clones are identified by sequencing across MMLV vector/genomic DNA junction sites. Importantly, gene-trap studies can identify genes important for any step in the replication cycle, do not require prior knowledge of the gene, and the roles of a prioritized subset of all human genes in viral infection can be confirmed independently by RNA interference (RNAi).
Lytic viral selection has been employed previously to identify HIV-1-dependency factors in Jurkat T cells whose silencing conferred survival in a genome-wide shRNA screen. 14 In our experimental system using HeLa cell-derived gene-trap libraries, the maximum CPE observed following HIV-1 infection was ∼90% (MOI=100), which was an unacceptably high background of surviving cells to isolate resistant clones. However, as unrelated lytic viruses show conserved utilization of cellular genes, 19,20 we tested 87 disrupted genes identified in cowpox, Ebola, influenza A, Marburg, and reovirus gene-trapping studies for conserved roles in HIV-1 replication. An additional seven targets were secondarily selected for investigation in light of their known pathway associations with critical trapped targets. In siRNA validation screens in an immortalized cell line, we identified several novel host genes, which support HIV-1 replication and other viruses. The roles of critical genes in HIV-1 replication were confirmed in primary human macrophages, and evidence is presented that most of these proteins may facilitate replicative steps prior to or including transactivator of transcription (Tat)-mediated gene transcription. Bioinformatics analyses suggested that many of the gene products participate in conserved pathways or functions important for HIV-1 replication, including gene transcription, signal transduction, mRNA splicing, protein ubiquitination, vesicular transport, and autophagy.
Materials and Methods
Cell culture
Primary macrophages were purified from fresh peripheral blood mononuclear cells by adherence to plastic tissue culture dishes, as described. 22 Using this method, we previously showed purity of isolated mononuclear phagocytes of >95% by flow cytometry. 23 Briefly, primary monocyte-derived macrophages were isolated from healthy HIV-1-negative blood donors by Ficoll-Hypaque centrifugation followed by adherence for 7 days to plastic Petri dishes. During differentiation, macrophages were cultured in Iscove's modified Dulbecco's medium supplemented with 20% fetal calf serum and 10% of human AB serum.
The following reagent was obtained through the NIH AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH: TZM-bl from Dr. John C. Kappes, Dr. Xiaoyun Wu, and Tranzyme Inc. U373-MAGI-CCR5 cells, obtained through the NIH AIDS Research and Reference Reagent Program (contributed by Dr. Michael Emerman and Dr. Adam Geballe), are a cell line derived from a glioblastoma that has been modified by stable transfection of expression vectors encoding LTR-β-galactosidase and CCR5 to enable infection by HIV-1. 24
Gene-trap library construction
The U3neoSV1 retrovirus shuttle vector 25 was obtained from H. Earl Ruley (Vanderbilt University). RIE-1, MDCK, and Vero E6 cells served as suitable parental cell lines for the preparation of gene-trap libraries, as they are efficiently killed by infection with reovirus (RIE-1 cells), influenza A (MDCK cells), and either cowpox, Ebola, or Marburg viruses (Vero E6 cells). RIE-1, MDCK, and Vero E6 gene-trap libraries were prepared as described. 19 –21
Generation of cell lines resistant to lytic viral infection from gene-trap libraries
Clonal reovirus, Ebola, and Marburg virus-resistant cell lines were derived from parental RIE-1 (reovirus, Lang strain) and Vero E6 (Ebola, 1976 Mayinga strain and Marburg, 1967 Voege strain) gene-trap library cells as described. 19 –21 Vero E6 library cells were also used to select cell lines surviving cowpox infection (Brighton strain), and MDCK library cells were used for influenza (A/PR/8/34 strain) studies. Briefly, gene-trap libraries, each harboring approximately 104 gene entrapment events, were expanded to 80–90% confluency until approximately 103 daughter cells represented each clone. Vero E6 cells were infected with cowpox using a multiplicity of infection (MOI) of 0.01, and MDCK cells were infected with influenza A using an MOI of 0.1. Infection proceeded at 37°C until >90% of cells were dead, at which point the medium was changed every 2–3 days to remove dead cells. Dead cells were removed by aspiration for 2–3 weeks until surviving clones were visually observed. Individual clones were detached with trypsin and isolated into separate wells of 24-well plates. Following expansion, resistance confirmation was performed by replica plating clones into duplicate 24-well plates, and reinfecting clonal cell lines with a 20-fold higher MOI than used in the initial viral selection for 2 h at 37°C before changing the medium. Clones that showed >70% survival following reinfection were selected for expansion to identify trapped genes.
U3neoSV1 shuttle vector rescue and sequencing
Genomic DNAs from clonal virus-resistant cell lines were extracted using a QIAamp DNA Blood Mini kit (QIAGEN, Inc., Valencia, CA). Shuttle vectors and genomic DNA flanking the U3neoSV1 integration site were recovered by restriction enzyme digests of genomic DNA, self-ligation, transformation into Escherichia coli, and sequencing the resultant carbenicillin-resistant plasmids to identify trapped genes, as described. 19 –21
Viruses and antibodies
The following HIV-1 strains were purchased from the Virology Core Facility, Center for AIDS Research at Baylor College of Medicine, Houston, TX: M-tropic HIV-1SF162 and HIV-1ADA, dual-tropic HIV-189.6, which is an HIV-1 laboratory adapted strain originally isolated from infected individuals. HIV-1SX is a chimeric M-tropic virus encoding the majority of the HIV-1JRFL envelope protein in an HIV-1NL4-3 backbone. Antibodies for detecting RAB9A, CCR5, DHX8, KALRN, UBA3, and HAP1 protein expression in U373 cell lines and macrophages were obtained from Abcam Inc. An anti-RAB11A antibody was obtained from Invitrogen. Antibodies recognizing CD4, ERBB2IP, VMP1, and UBE2E3 were obtained from Abnova. An anti-β-actin antibody was obtained from Santa Cruz Biotechnology. These antibodies were used to perform Western dot-blot analysis.
Western dot-blot analysis
Cellular expression and downregulation of cellular proteins were analyzed in Western dot-blot analysis using primary antibodies specific for cellular proteins and secondary antihuman antibody conjugated to HRP (obtained from Santa Cruz Biotechnology).
siRNA transfections
siRNA SMARTpools were obtained from Dharmacon. TZM-bl cells were transfected with siRNAs as described. 19 Primary human macrophages were transfected with siRNAs using oligofectamine (Invitrogen) according to the manufacturer's instructions. Primary macrophages adherent for 5 days were seeded overnight in 96-well plate format (1×105 cells / well) and subsequently transfected with 50 nM siRNA in serum-free Iscove's medium. Cells were fed with Iscove's medium and 20% fetal calf serum (FCS) after 4 h to terminate transfection. To measure protein expression following RNAi, cells were lysed at 48 h posttransfection in phosphate-buffered saline (PBS) with 1% Triton X-100 in preparation for Western dot-blot analysis. U373-MAGI-CCR5 cells were transfected with 50 nM siRNA 24 h after plating at 1×104 cells per well in 96-well microtiter plates. Protein expression and downregulation in U373-MAGI-CCR5 cells were assessed at 48 h post-siRNA transfection by lysing cells in 1% Triton X-100 in PBS and detecting target proteins by Western dot-blot analysis.
Real-time semiquantitative PCR
Total mRNA was isolated from parental and siRNA-transfected cells using the RNeasy Kit (Qiagen, Inc.), and reverse transcribed using random hexamers (Applied Biosystems). Real time PCR was performed as described 26 using an Mx4000 Multiplex Quantitative PCR System (Stratagene). Real time PCR detection assays for all siRNA target genes described in this study were from Applied Biosystems. Target gene mRNA expression levels were normalized to hypoxanthine-guanine phosphoribosyltransferase (HGPRT) mRNA expression levels, using the human HGPRT TaqMan assay kit (Applied Biosystems).
HIV-1 p24 and β-galactosidase assays
HIV-1 p24 ELISA assays were used to assess HIV-1 production in TZM-bl cells and primary human macrophages, and β-galactosidase assays measuring HIV-1 Tat-dependent gene expression were performed in U373-MAGI-CCR5 cells. HIV-1 p24 assays were performed in TZM-bl cells as described previously. 19 Primary macrophages adherent for 7 days were seeded overnight in 96-well plates (1×105 cells per well) and transfected after 24 h with 50 nM siRNAs for 48 h. Subsequently, macrophages were infected for 2 h with either SF162, ADA, or 89.6 HIV-1 strains (MOI=0.02), after which the medium was changed to remove the inoculum. Cells were fed with fresh medium on day 4 postinoculation, and supernatants were assessed for virus p24 production using the Antigen Capture ELISA assay (ImmunoDiagnostics) on day 7 postinoculation.
U373-MAGI-CCR5 cells express β-galactosidase under the control of the HIV-1 LTR, which is transactivated by the HIV-1 Tat protein. U373-MAGI-CCR5 cells (1×104/well) were plated in 96-well microtiter plates 12 h before siRNA transfections. At 48 h posttransfection the cells were infected with HIV-1 strains (MOI=0.02). After 2 h postinoculation at 37°C, the medium was changed and β-galactosidase activity was measured 48 h after infection using the Beta-Glo Assay System (Promega).
Cytotoxicity assays
Cytotoxicity of siRNA was measured using the aCella-Tox Bioluminescence Cytotoxicity assay kit according to the protocol provided by the manufacturer (Cell Technology Inc., Mountain View, CA). This assay detects secreted glyceraldehyde-3-phosphate dehydrogenase (GAPDH) in cells with compromised membrane integrity. Cell supernatants were harvested at 48 h posttransfection and GAPDH enzyme activity was measured by luminometry.
Bioinformatics analyses
Eight genes implicated in HIV-1 replication during siRNA screens (DHX8, GTF2E1, GTF2E2, HAP1, KALRN, UBA3, UBE2E3, and VMP1) were analyzed using Ingenuity Pathway Analysis (IPA, Ingenuity Systems, Inc.). To allow comparison with prior genome-wide siRNA screens by Brass et al., 16 Konig et al., 11 Zhou et al., 15 Yeung et al., 14 and Liu et al., 27 as well as a systematic affinity tagging mass spectrometry study by Jäger et al., 28 additional IPA analyses were performed on their 281, 292, 232, 252, 114, and 497 gene datasets, respectively. These analyses identified canonical pathways from the IPA library of canonical pathways that were statistically significant compared to the datasets from each study, based on Fisher's exact test. Canonical pathways with a calculated p-value less than 0.01 were considered significant, implying that the probability of a chance association between genes in the dataset and the canonical pathway is less than 1%. Functional analyses were also performed with the above datasets, identifying biological functions that were statistically significant with functions in the Ingenuity Knowledge Base based on the right-tailed Fisher's exact test (p-value<0.05).
Results
Identification of cellular genes facilitating infection by gene-trap mutagenesis
Ongoing gene-trap studies in our collaborative laboratories with multiple viruses and bacteria/bacterial toxins have identified candidate genes that may be critical for cytotoxic infection and/or intoxication. 18 –21,29,30 Here we show HIV-1 siRNA-screening data for genes disrupted in clonal cell lines surviving infection with one or more of several lytic viruses studied. Table 1 lists 87genes that were disrupted in clonal cell lines surviving lytic infection with either cowpox virus, Ebola virus, influenza A virus, Marburg virus, or reovirus, and seven nontrapped genes associated with trapped gene pathways later found critical for HIV-1 replication. Candidate genes participate in virtually all aspects of cellular physiology, with ∼50% of the genes functioning in transcription, signal transduction, trafficking, nucleic acid metabolism, or as metabolic enzymes. Additional cellular roles essential for viral replication were associated with protein translation and ubiquitination, autophagy, chaperone and kinase functions, cell–cell adhesion, cell cycle progression, proliferation, and cytoskeletal regulation.
Host genes identified by gene-trap insertional mutagenesis studies with either cowpox virus, Ebola virus, influenza A virus, Marburg virus, or reovirus were screened for potential roles in HIV-1 replication using small interfering RNAs (siRNAs) to silence target gene expression prior to performing HIV p24 assays. Additional nontrapped genes included as candidates in siRNA screens were selected based on known associations with trapped genes. Genes important for HIV-1 replication are shown in bold.
siRNA validation screening of trapped genes in immortalized cells
RNAi screens were performed in CD4+/CCR5+/CXCR4+ TZM-bl HeLa cells with the X4-tropic HIV-1 strain LAV (Fig. 1). We previously provided evidence that RAB9Aand Rab11A support viral egress from late endosomes to plasma membranes, and that ADAM10 regulates a critical step in trafficking viral cDNA to the nuclear compartment.
18,19
Thus, RAB9A
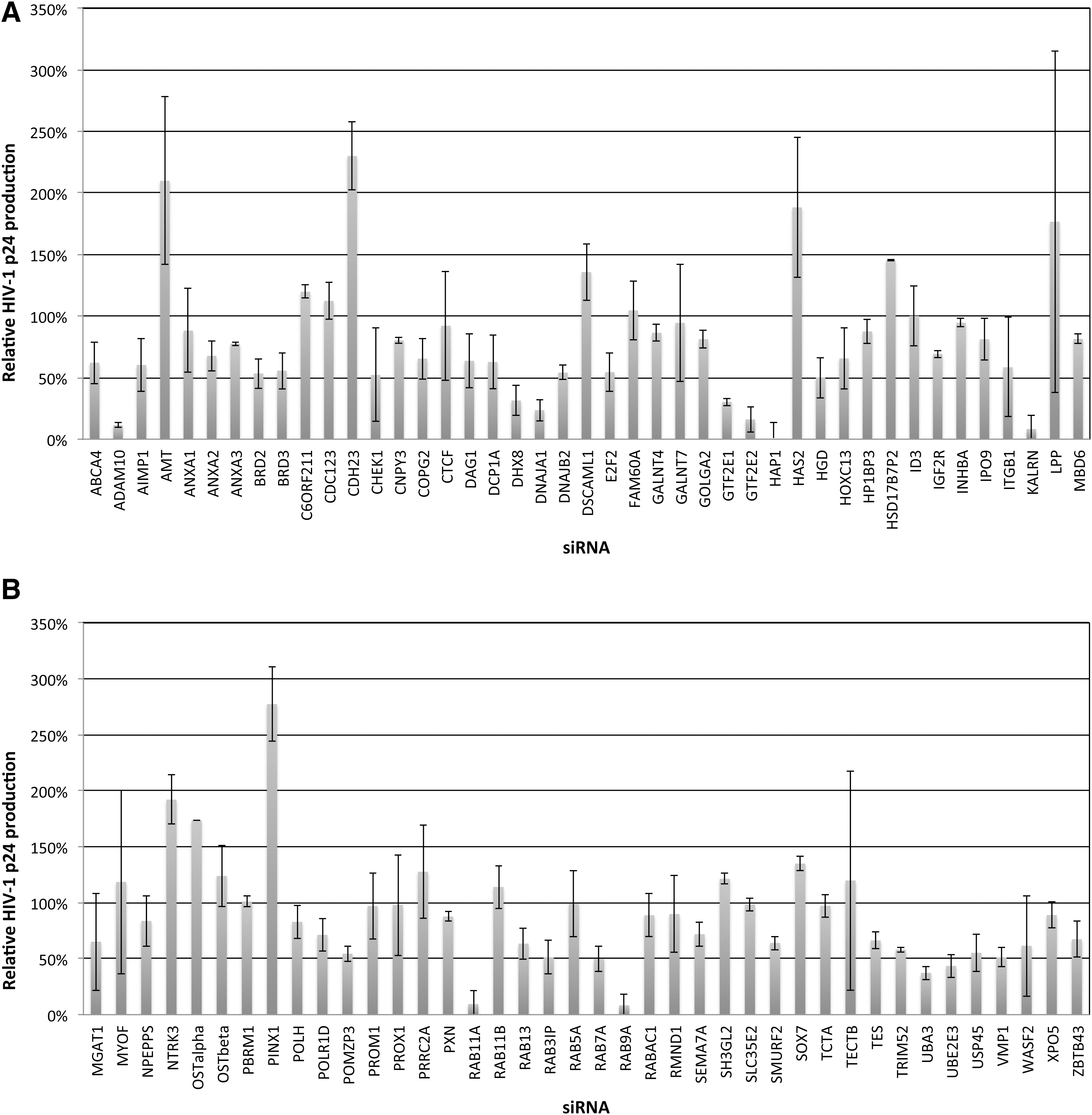
Small interfering RNA (siRNA) screening of candidate genes identified by gene trap insertional mutagenesis or association with critical trapped gene pathways in HIV p24 assays. Candidate trapped genes were identified in clonal cell lines resistant to lytic infection in studies with cowpox virus, Ebola virus, influenza A virus, Marburg virus, or reovirus. To test candidate genes for potential roles in supporting HIV replication, TZM-bl HeLa cells were transfected in T25 flasks with the indicated siRNAs (50 nM) for 48 h prior to infection with HIV (X4-tropic LAV strain, MOI=1). Following infection, cells were detached, washed in PBS, and expanded in duplicate T75 flasks for 3 days. HIV replication was assayed by measuring p24 secretion into culture supernatants in HIV p24 ELISA assays.
A complex containing the viral Tat protein, the cellular RNA polymerase II, and several associated general transcription factors (TFIIs) initiate HIV-1 gene transcription. 35,36 Although several TFIIs have been shown to stimulate HIV-1 transcription including TFIID, TFIIF, TFIIH, and TFII-I, 37 –40 the importance of TFIIE has not been demonstrated. We found that both the α and β subunits of TFIIE (GTF2E1 and GTF2E2, respectively) support replication with R5-, R5X4-, and X4-tropic strains in TZM-bl cells (Figs. 1A and 2A). ERBB2IP siRNA was used as a negative control, as silencing expression of this gene does not affect HIV-1 replication. 18 GTF2E1 silencing also inhibited Tat-dependent gene expression in U373-MAGI-CCR5 cells encoding a stably integrated β-galactosidase (β-gal) reporter gene under the control of the HIV-1 long terminal repeat (Fig. 2D). GTF2E1 or GTF2E2 siRNAs efficiently silenced expression of the respective mRNAs in TZM-bl (Fig. 2B and C) and in U373-MAGI-CCR5 cells (Fig. 2E), as determined by real time PCR analysis with normalization to HGPRT expression.
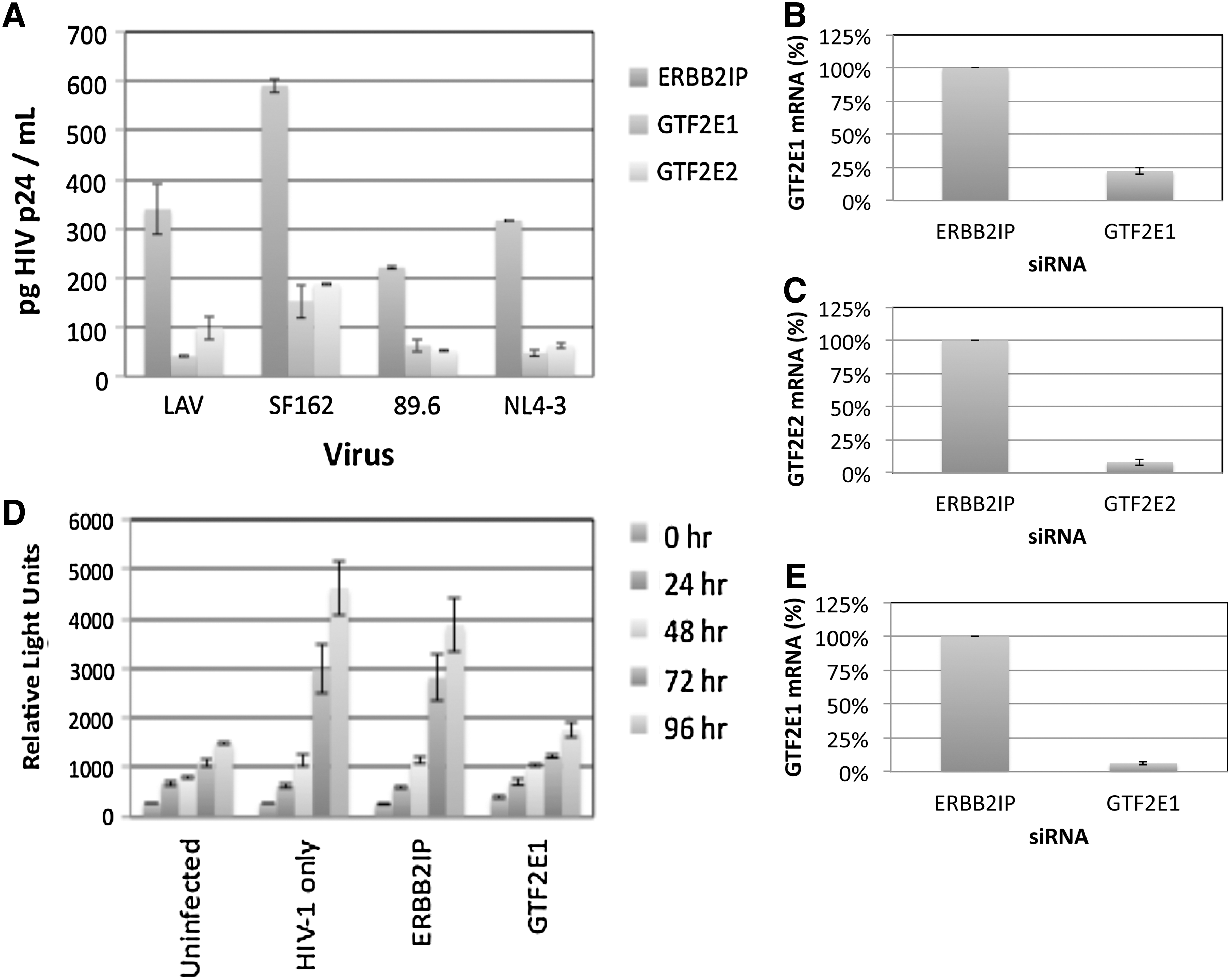
Dependence of HIV-1 replication and Tat activity on host cell general transcription factor IIE.
Expression of critical host factors and silencing by siRNA
RNAi screens performed in TZM-bl cells revealed several candidate genes whose expression appeared critical for HIV-1 replication (Fig. 1). DHX8, HAP1, KALRN, RAB9A,RAB11A, UBA3,UBE2E3, and VMP1 were selected for further study in primary human macrophages to confirm their roles in more physiologically relevant cells, and in U373-MAGI-CCR5 cells to test their requirements in Tat-dependent gene expression. As DNAJA1 has been previously characterized to be critical for HIV-1 replication, 33 it was not studied further. Western dot-blots of the proteins encoded by these candidate genes were used to demonstrate that each of the indicated proteins was expressed in both primary macrophages and U373-MAGI-CCR5 cells, and that siRNAs downregulated target proteins by ∼80–90% (Fig. 3A and B).
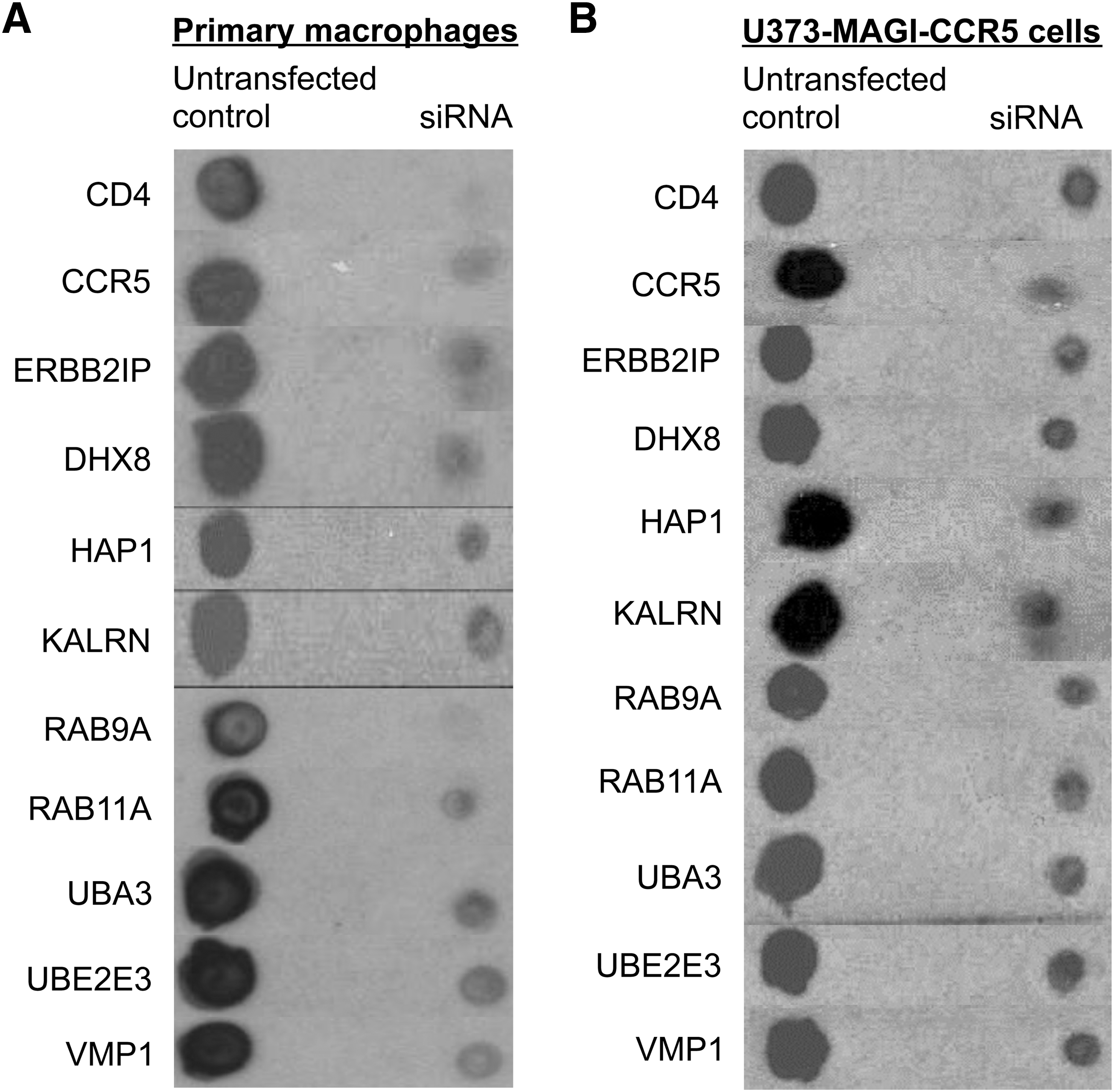
Down-regulation of critical host proteins in primary human macrophages and U373-MAGI-CCR5 cells. Untransfected or siRNA-transfected
Role for critical host cell factors in HIV-1 replication in macrophages
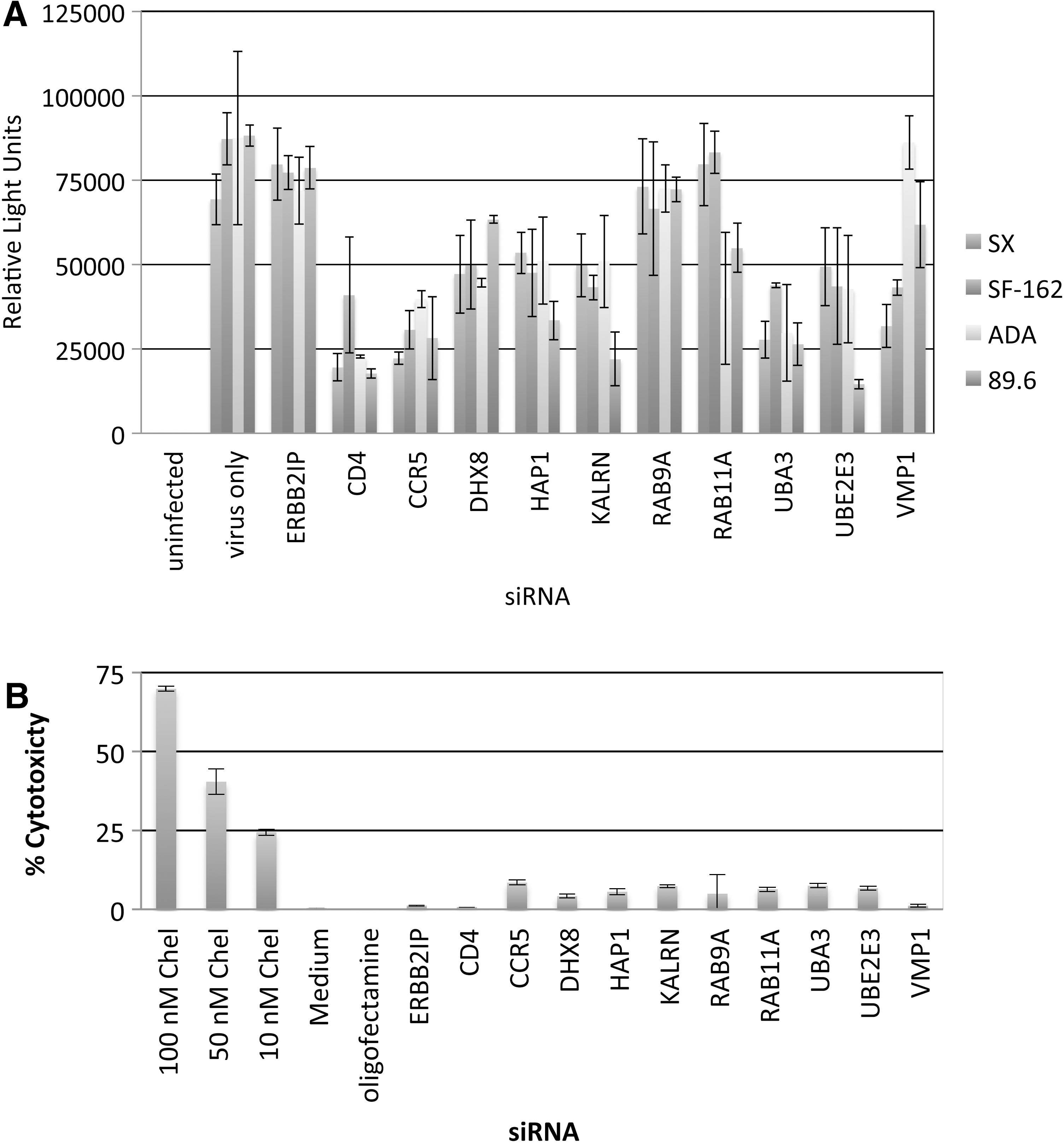
Candidate genes were silenced with siRNAs to confirm their roles in supporting HIV-1 infection in primary human macrophages (Fig. 4). Controls consisted of primary macrophages transfected for 48 h with siRNAs directed against ERBB2IP (negative control) and CD4 or CCR5 (positive controls; major receptor and coreceptor for R5-tropic HIV-1). Primary macrophages were similarly transfected with siRNAs targeting candidate genes prior to infection with R5-tropic (SF162 or ADA) or R5X4-tropic (89.6) strains (Fig. 4A–C). ERBB2IP siRNA did not inhibit viral p24 production relative to nontransfected cells. However, replication of SF162, ADA, and 89.6 HIV-1 strains was dramatically reduced by silencing candidate genes, with inhibition in most cases being comparable to CD4 and CCR5 siRNA positive controls. These results independently confirm the importance of the indicated cellular proteins in supporting HIV-1 replication in primary macrophages with three different strains.
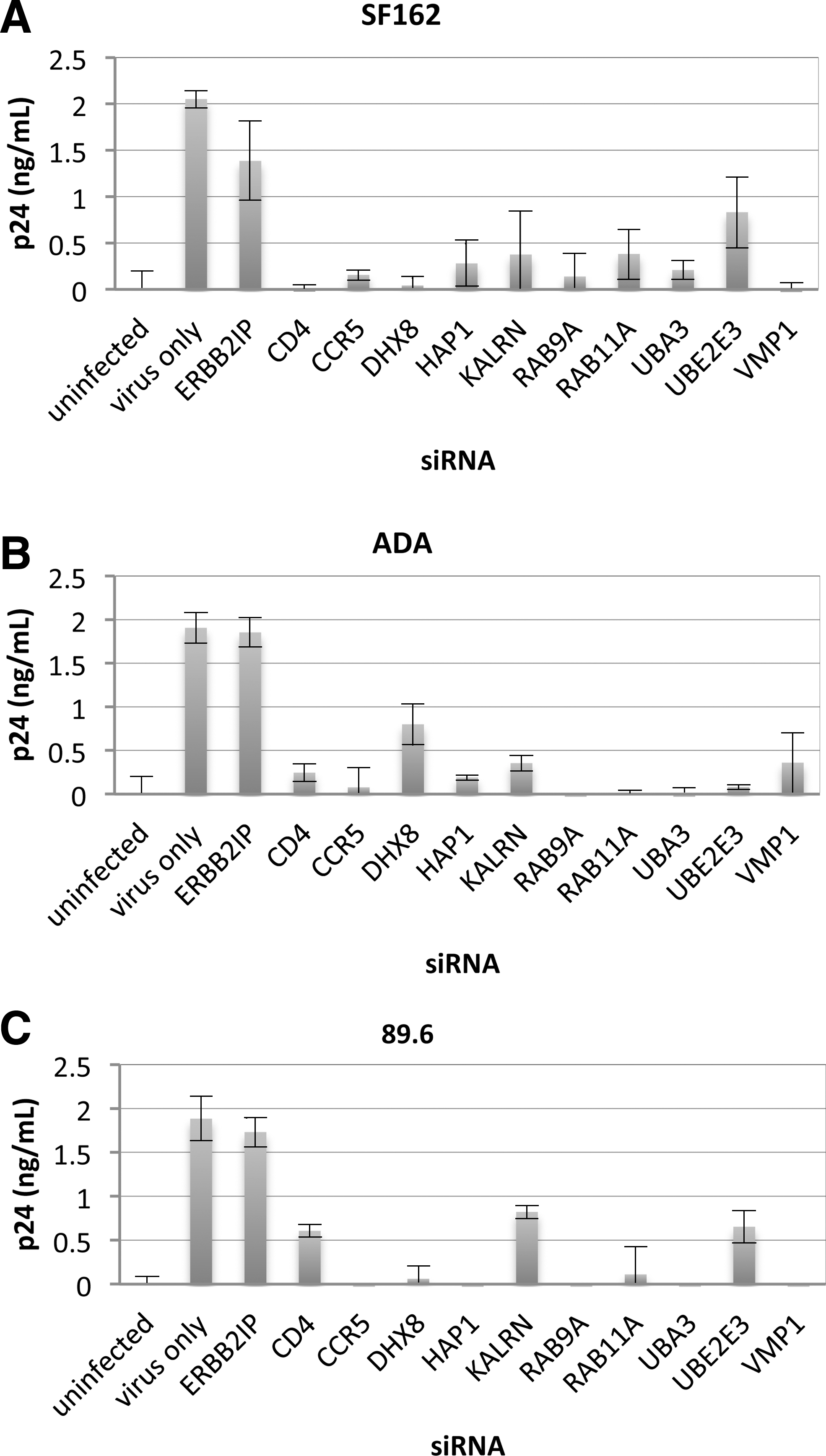
Inhibition of HIV-1 infection with R5 virus strains SF162 and ADA, and dual-tropic 89.6 following target gene silencing in primary human macrophages. Monocyte-derived macrophages were purified by adherence to plastic and transfected with 50 nM siRNAs after 5 days of adhesion to tissue culture plates. Following a 48-h transfection period, macrophages were infected (MOI=0.02) with
Assessing early stage activity of host genes in HIV-1 replication
To determine whether targeted genes support early replication steps (up to and including HIV-1 gene expression), U373-MAGI-CCR5 β-gal reporter cells were transfected with control siRNAs or siRNAs targeting DHX8, HAP1, KALRN, RAB9A, RAB11A, UBA3, UBE2E3, or VMP1, and subsequently studied in Tat-dependent β-gal-based infectivity assays. At 48 h posttransfection, U373-MAGI-CCR5 cells were inoculated with R5-tropic (SX, ADA, and SF162) or R5X4-tropic (89.6) HIV-1 strains. Subsequently, cells were lysed and the effects of target gene silencing on Tat activity was assessed by measuring β-gal activity as relative light units, using the Beta-Glo Assay (Promega). Tat-dependent β-gal expression was not inhibited by silencing ERBB2IP, whereas silencing CD4 and CCR5 reduced β-gal activity in cells infected with either R5-tropic HIV-1 strains (SX, SF162, or ADA) or the dual tropic strain 89.6 (Fig. 5A). RAB9A and (in 2/4 cases) RAB11A silencing did not inhibit Tat-dependent gene expression, consistent with their proposed roles in regulating vesicular transport of translated viral proteins as a precursor to assembly/budding, occurring downstream of viral gene expression. 19,41,42 With the exception of VMP1, silencing the other candidate genes consistently inhibited Tat-dependent gene expression with each of the four HIV-1 strains tested. These data suggest a role for RAB9A and RAB11A at posttranslational steps in the HIV-1 replication cycle, and indicate that the other selected proteins may function at steps prior to Tat-dependent HIV-1 transcription.
Effects of candidate gene silencing on Tat-dependent gene expression and cell viability in U373-MAGI-CCR5 β-gal reporter cells.
Variability in inhibition of Tat-dependent gene expression following target gene silencing was observed between the four HIV-1 strains tested. To determine whether Tat-dependent gene expression and/or the observed interstrain variability might result from transfection toxicity, candidate genes were silenced in U373-MAGI-CCR5 cells and studied using the aCella-Tox Bioluminescence Cytotoxicity assay (Fig. 5B). Cytotoxicity is proportional to measurable GAPDH activity in culture supernatants, resulting from lost membrane integrity. Minimal toxicity (<10%) was observed in siRNA transfectants, whereas the positive control chelerythrine (a cell-permeable protein kinase C inhibitor) showed a dose-dependent increases in cellular toxicity ranging from 25% (10 nM) to 70% (100 nM).
Bioinformatics analyses of host factors supporting HIV-1
Host genes found critical for efficient HIV-1 replication in this study (DHX8, GTF2E1, GTF2E2, HAP1, KALRN, UBA3, UBE2E3, and VMP1) were compared with genes implicated in four prior genome-wide siRNA screens against HIV-111,14–16 and a recent systematic affinity tagging mass spectrometry study for HIV-human protein–protein interactions 28 using Ingenuity Pathway Analysis software (IPA, Ingenuity Systems, Inc.). None of the eight genes identified in this study matched genes found in the five prior studies, although this might not be surprising given the low overlap between the studies. 9,28,43 However, meta-analyses have shown a greater degree of overlap in biological pathways and functions associated with productive infection than in exactly matching genes between studies. 9,14,43 Target genes identified in this study participate in seven canonical pathways and 12 biological functions that were significantly represented in the five previous studies (Tables 2 and 3). In some cases, the same genes facilitate more than one pathway or function. Thus, the eight genes identified here are novel targets consistent with biological pathways and functions previously found to be utilized by HIV-1 by both siRNA- and proteomics-based screens.
p<0.01 by Fisher's exact test.
DHX8, GTF2E1, GTF2E2, HAP1, KALRN, UBA3, UBE2E3, and VMP1 were analyzed by IPA software to determine which of their associated canonical pathways overlapped with significant associated pathways implicated in four previous siRNA screens against HIV-1, 11,14 –16 and a systematic affinity tagging/purification mass spectrometry study. 28
p<0.05 by right-tailed Fisher's exact test.
We also performed IPA analysis to study genes whose silencing resulted in modest increases in HIV-1 replication (Fig. 1: AMT, CDH23, HAS2, LPP, NTRK3, OSTα, and PINX1), comparing them to 114 HIV-1 restriction factors identified by Liu et al. 27 None of the indicated genes was found among the 114 restriction factors, or in their significantly associated biological pathways or functions. This finding is in agreement with the nature of the gene-trap selection process used, as survival of mutant cells from a lytic infection favor limitation of virus replication.
Discussion
We used a forward selective approach to discover cellular genes participating in the replication of several pathogenic viruses, and assayed candidate genes for functional roles in HIV-1 infection. Many of the trapped genes are related to genes identified in previous studies with other viruses by virtue of their being putative binding partners, regulating the same pathways, or sharing identity. However, with the exception of DNAJA1, 33 none of the genes identified as critical for HIV-1 in this study has been previously implicated with HIV-1. Cellular genes were initially tested in p24 ELISA assays to reveal targets whose downregulation may compromise any step in the replication cycle, and subsequent β-gal assays indicated whether they regulate steps prior to or during Tat-dependent gene expression, or subsequent steps.
This study found several novel genes whose products augment HIV-1 replication. Moreover, it highlights known biological functions of these genes, which may relate to the viral life cycle. Of these, both subunits of TFIIE (GTF2E1 and GTF2E2) were critical for the replication of four different HIV-1 strains (Fig. 2A); furthermore, GTF2E1 silencing inhibited Tat-dependent gene transcription (Fig. 2D). In addition to the known role of TFIIE in the assembly of RNA polymerase II complexes, 44 IPA analysis also indicated that GTF2E1/GTF2E2 may be important in estrogen receptor, glucocorticoid receptor, and androgen signal transduction pathways associated with canonical pathways implicated in previous siRNA screens (Table 2). In gene-trap studies, a single GTF2E1 allele was disrupted in RIE-1 cells surviving lytic reovirus selection without any detriment to host cell viability.
Following gene transcription, viral pre-mRNAs are spliced and shortened mRNAs encoding regulatory proteins are exported to the cytoplasm. DHX8 is a cellular RNA helicase that regulates the release of spliced mRNA from spliceosomes in preparation for nuclear export, 45 which may explain its requirement in HIV-1 production at a step prior to Tat-dependent gene expression (Figs. 1A, 4, 5, and Table 3). DHX8 is also putatively involved in influenza A virus replication, as determined by siRNA screening in a human osteosarcoma cell line. 46
Viral assembly can initiate on late endosomes, where ubiquitinated viral proteins recruit ESCRT complexes that direct subsequent budding. 47 –49 As a ubiquitin-conjugating enzyme, UBE2E3 (Table 2) potentially modifies HIV-1 proteins, whereas HAP1 (Table 3) and KALRN may regulate viral protein trafficking to late endosomes and/or HRS recruitment. 50 –52 UBA3 is a member of the E1 ubiquitin-activating enzyme family and can serve in complex with amyloid precursor protein binding protein-1 to posttranslationally modify Cullin 4a through conjugation of NEDD8 (Table 3), a small ubiquitin-like protein. 53 NEDD8 conjugation of the Cullin subunit of Cullin-RING E3 ubiquitin ligases is thought to stimulate substrate polyubiquitination. 54 These observations could account for the utilization of UBA3, UBE2E3, HAP1, and KALRN during viral replication (Fig. 1), but does not explain their observed requirement to complete viral replication steps prior to Tat-dependent gene expression (Fig. 5A). Ubiquitination often marks cellular proteins for lysosomal degradation. Interestingly, Tat ubiquitination actually enhances its activity through a nonproteolytic mechanism. 55 Thus, UBA3, UBE2E3, KALRN, and HAP1 may potentiate HIV-1 replication during early and/or late stages in the viral life cycle. RAB9A and RAB11A were not required for Tat-dependent gene expression, consistent with their proposed roles in governing vesicular transport of viral proteins from late and recycling endosomes to plasma membranes in advance of egress. 19
In summary, we present 87 host genes whose disruption conferred resistance to lytic cowpox virus, Ebola virus, influenza A virus, Marburg virus, or reovirus infections in gene-trap insertional mutagenesis studies. RNAi screens performed in TZM-bl cells implicated six trapped genes and two associated cellular targets (GTF2E2 and HAP1) during X4-tropic LAV HIV-1 replication, and critical host genes were further validated in HIV-1 p24 assays in primary human macrophages and β-gal assays. siRNA knockdown of candidate genes in primary macrophages, infected with both R5-tropic viruses (SF162 and ADA) and a dual-tropic R5X4 strain (89.6), demonstrated the potential physiological relevance of identified genes in supporting replication of R5-, R5X4-, and X4-tropic strains. IPA analyses provided evidence that critical host factors identified in this study are members of significantly represented canonical pathways with putative functions previously associated with genes discovered in genome-wide screens against HIV-1, independent of whether the host factors were identified by targeted gene silencing or physical interactions with viral proteins. Further mechanistic studies are required to determine the precise roles of the gene products in HIV-1 replication and to develop novel compounds to inhibit their functions.
Footnotes
Acknowledgments
N.D., G.L., and M.R.F. were supported by the Public Health Service (PHS) Grant AI084705 from the National Institute of Health AIDS SBIR and partly supported by PHS HL088999 from the National Heart, Lung, and Blood Institute. D.H.R. and J.S. were supported by the PHS, and a gift from the Red and Bobby Buisson Foundation. The views expressed in the article do not necessarily represent the views of the VA.
Author Disclosure Statement
J.L.M., T.W.H., and W.A.O. were supported by Zirus, Inc., a biotechnology company dedicated to the discovery of new targets for antiviral therapeutics.