
Abstract
Thirty HIV-1 URF_01AE/ B′ complete or nearly full-length genome sequences sampled within Southeast Asia were obtained from the Los Alamos HIV Sequence Database. Phylogenetic and recombinant analyses revealed that three sequences indeed displayed the identical recombinant structure. Of note, the three subjects, harboring novel CRF01_AE/B recombinants, did not have apparent epidemiological linkage. They fulfilled the criteria for the designation of a new circulating recombinant form (CRF) and constituted the 52nd CRF identified in the worldwide HIV-1 pandemic. In this chimera, two short subtype B segments were inserted into a backbone of CRF_01AE. The breakpoints corresponded to HXB2 nucleotide positions 2930, 3251, 8521, and 9004 approximately. This CRF is the first one identified by neatening and analyzing the sequences already presented in the Los Alamos HIV Sequence Database. This indicates that we should pay attention not only to explicit subtype sequences but also to those classified as a unique recombinant form (URF) so far.
M
Southeast Asia was thought to be an epicenter of the HIV-1 epidemic in Asia. The AIDS epidemic in this region was initiated by two different subtype strains in two separated risk groups, with subtype B among injecting drug users (IDUs) and CRF01_AE among those heterosexually exposed, throughout the early phase. 2,3 CRF01_AE can clearly cross over from the heterosexual epidemic to IDUs and by 1995 it also became the predominant subtype in IDUs. 4 The cocirculation of the two different strains in this region led to the generation of more complex recombinant strains that emerged in almost every country in Southeast Asia. 5 –10 Furthermore, some of the recombinant strains have spread widely in populations and have become CRFs. Up to now, five CRFs, originated from CRF01_AE and subtype B′ lineages, have been reported in Southeast Asia: CRF15_01B 11 and CRF34_01B 12 from Thailand, CRF33_01B 13 and CRF48_01B 14 from Malaysia, and CRF51_01B 15 from Singapore. The HIV-1 epidemic in this region has now become increasingly heterogeneous.
The extensive use of complete and nearly full-length genome sequencing of HIV-1 strains has provided a powerful and accurate approach to the molecular epidemiology of regional epidemics. Consequently, the number of complete or nearly full-length HIV-1 genome sequences was growing larger and larger. The majority of these can be classified as subtypes and CRFs definitely, while the minority remains as URFs. To define a CRF, at least three epidemiologically unlinked HIV-1 sequences with identical mosaic structures should be characterized, at least two of them in the nearly full-length genome (8 kb)
1
. For the present study, we identified a novel CRF_AE/B′ candidate, which emerged in Southeast Asia, using sequences obtained from the Los Alamos HIV Sequence Database (
Thirty HIV-1 URF_01AE/ B′ complete or nearly full-length genome sequences sampled within Southeast Asia were obtained from the Los Alamos HIV Sequence Database. These sequences were generated by sequencing from plasma or peripheral blood mononuclear cell (PBMCs) samples collected from Thailand (n=19), Malaysia (n=8), Myanmar (n=2), and China (n=1). In addition to these 30 sequences, we also retrieved subtype reference sequences from the same database. All the sequences were incorporated into one sequence dataset using SynchAlign, softwarehttp://www.hiv.lanl.gov/content/sequence/SYNCH_ALIGNS/SynchAligns.html, and edited using BioEdit software. A phylogenetic tree was constructed by the neighbor-joining method based on Kimura's two-parameter distance matrix with 1000 bootstrap replicates using Mega 5.04. With respect to recombination analysis, RIP,
Using phylogenetic analysis, we found that three nearly full-length genome sequences from three patients formed a monophyletic branch supported by a high bootstrap value of 99%, located outside any HIV-1 subtypes and known CRFs in Southeast Asia (Fig. 1). Both RIP and jpHMM revealed that the three URFs nearly full-length genome sequences indeed displayed the identical recombinant structure composed of CRF_01AE and subtype B. In this chimera, two short subtype B segments were inserted into a backbone of CRF_01AE. The breakpoints corresponded to HXB2 nucleotide positions 2930, 3251, 8521, and 9004 approximately (Fig. 2).
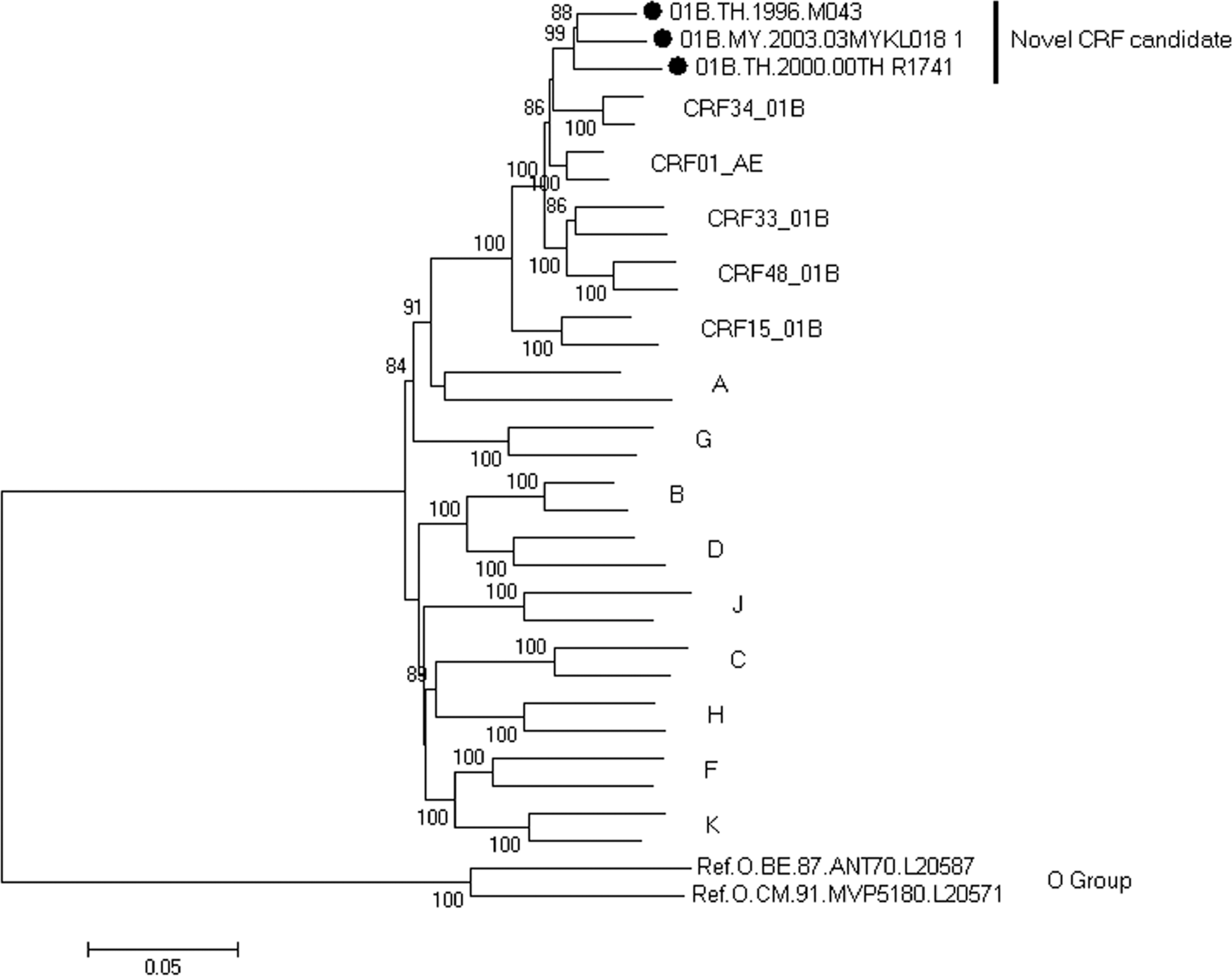
Identification of a novel circulating recombinant form (CRF) candidate. The neighbor-joining phylogenetic tree is constructed based on the nucleotide sequence of nearly full-length genomes using MEGA 5.04. The tree is rooted with group O. All the reference strains were retrieved from the Los Alamos HIV Sequence Database. Values on the branches represent the percentage of 1000 bootstrap replicates and bootstrap values over 70% are shown in the tree. The novel CRF candidate is marked with a solid circle. The scale bar indicates 5% nucleotide sequence divergence.
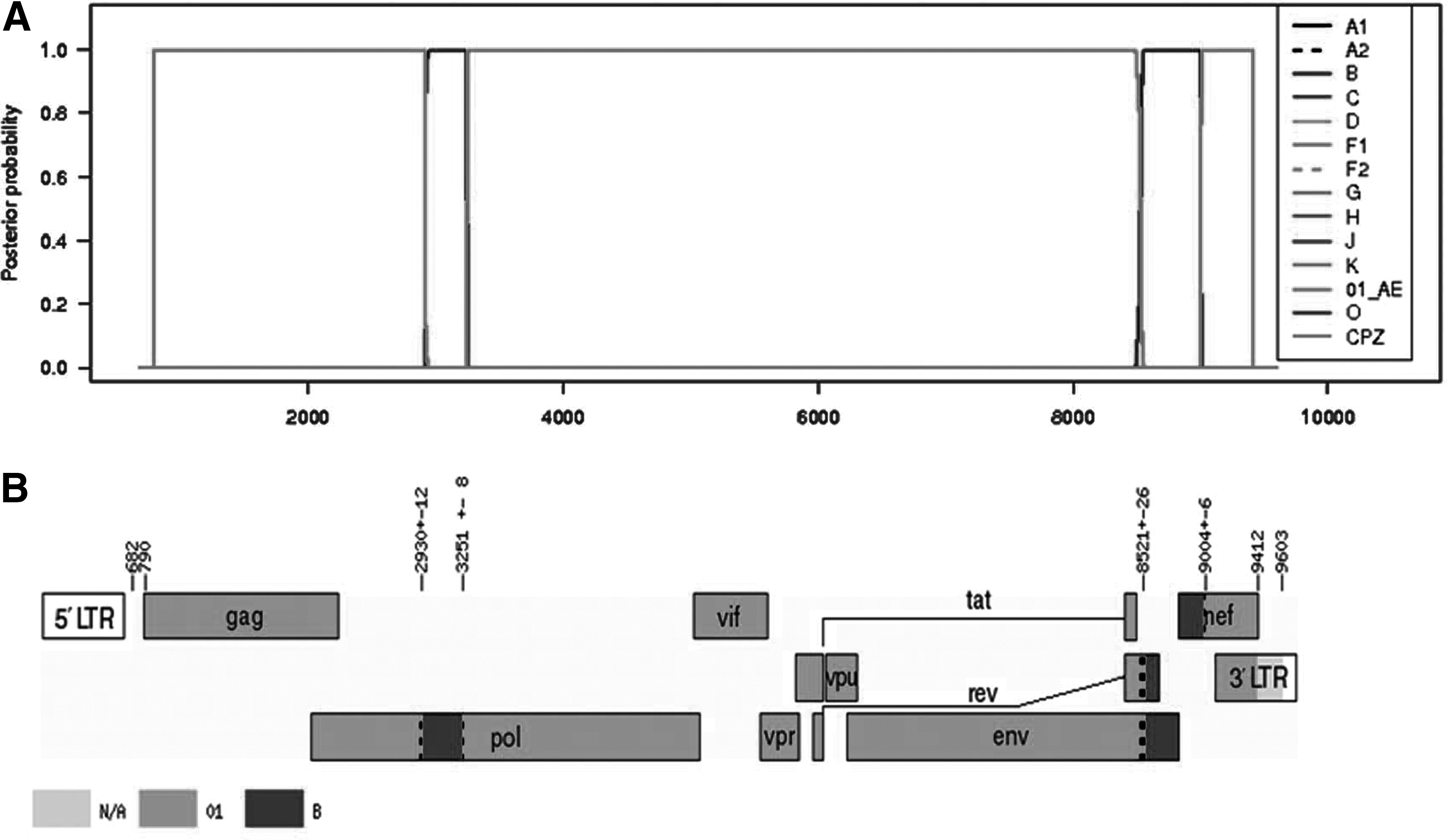
Recombinant analysis of the novel identified CRF52_01B.
To confirm the subtype structure of CRF52_01B and to estimate likely parental lineages, we divided the HIV-1 genomes into five segments according to the breakpoints. Subregion phylogenetic analysis confirmed the four breakpoints identified using jpHMM. The genomic segments I, III, and V clustered with reference subtype CRF01_AE while the other two segments II and IV branched with reference subtype B (Fig. 3). As shown in Fig. 3, the subregion tree analysis demonstrated that CRF01_AE segments (I, III, and V) belonged to the cluster of Thailand CRF01_AE other than African CRF01_AE, showing that the three CRF01_AE segments indeed originated from CRF01_AE of Thailand origin. Similarly, subtype B segment IV analysis revealed that it originated from Thailand B rather than Western B. However, we could not determine the origin of the remaining subtype B segment II because the length was too short to obtain high bootstrap values.
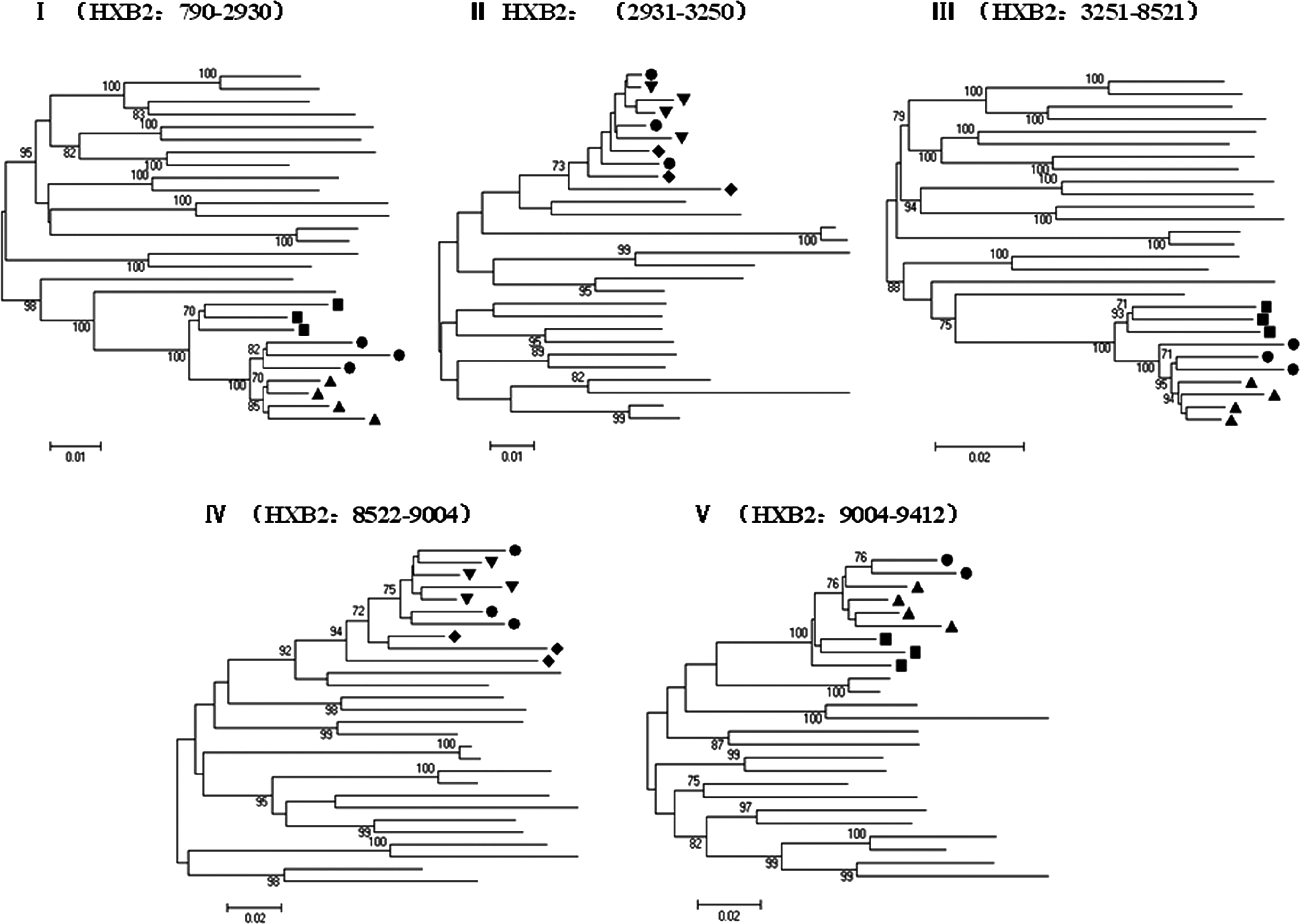
Phylogenetic trees for genome segments of the novel identified CRF52_01B. The neighbor-joining tree is constructed using MEGA 5.04.Values at the nodes indicate the percent bootstraps in which the cluster to the right was supported. Bootstraps of 70% and higher only are shown. The scale bars are shown at the bottom. •=CRF52_01B; ▴=Thailand CRF01_AE; ▪=African CRF01_AE;▾=Thailand B; □=Western B.
The profiles of the subtype structure of the three sequences were distinct from the other five CRFs originated from CRF01_AE and subtype B′ reported in Southeast Asia. Of note, the three nearly full-length genome sequences (01B.MY.2003.03MYKL018_1, 01B.TH.2000.00TH_R1741, and 01B.TH.1996.M043) were presented in three different studies 13,16,17 from three different patients (Table 1) with no apparent epidemiological linkage. These data established that the three sequences show the same recombinant structure over the whole genome, fulfilling the criteria for designation of a new CRF. They constitute the 52nd CRF identified in the worldwide HIV-1 pandemic, herein called CRF52_01B.
n/a, not available.
Detailed information about the three subjects harboring CRF52_01B recombinants is shown in Table 1. Unlike other CRFs identified worldwide, the most interesting element is that the three subjects belonged to two different countries, with two in Thailand and one in Malaysia, within Southeast Asia. However, the subjects were reported by year of first positive HIV-1 test, although that of subject M043 was not available. From Table 1, it is apparently that subject M043 was infected in or before 1996. The obviously wide geographic separation and early infection date of the three subjects suggested that CRF52_01B arose at least 15 years ago. To obtain accurate estimates regarding the time of origin and transmission direction of the novel CRF in Southeast Asia, more sequences with different locations and sampling times are needed in Bayesian coalescent analysis using BEAST software. 18,19
All of the three subjects reported sexual exposure, with two being heterosexual and one bisexual. It has been reported that most women infected in Southeast Asia have been the monogamous wives or regular partners of higher risk men. 20 Of interest, subject R1741 was one of the participants recruited from public family planning clinics in Thailand. Her only reported risk factor for HIV-1 infection was heterosexual exposure. 16 Taking subject R1741 into account, she should have acquired the HIV-1 infection from her male sexual partner. It was suggested that the novel CRFs have spread into low-risk people early.
In this study, we identified a novel CRF (CRF52_01B) composed of CRF01_AE and subtype B in Southeast Asia by analyzing the 30 complete or nearly full-length genome sequences obtained from the Los Alamos HIV Sequence Database. CRF52_01B is the sixth identified CRF that has originated from CRF01_AE and subtype B lineages within Southeast Asia in the past several years. What is most interesting is that this CRF is the first one identified by neatening and analyzing the sequences already presented in the Los Alamos HIV Sequence Database. This emphasizes that we should pay attention not only to explicit subtype sequences but also to those classified as URFs so far. The prevalence and public significance of the novel CRF remain to be elucidated in the future.
Footnotes
Acknowledgments
This work was supported by the National Key S&T Special Projects on Major Infectious Diseases (Grants 2008ZX10001-004 and 2008ZX10001-012).
Author Disclosure Statement
No competing financial interests exist.