
Abstract
Untreated HIV infection is associated with endothelial dysfunction and subsequent cardiovascular disease, likely due to both direct effects of the virus and to indirect effects of systemic inflammation on the vasculature. We have recently shown that treatment with the antiinflammatory agent pentoxifylline (PTX) improved in vivo endothelial function and reduced circulating levels of the inflammatory markers vascular cell adhesion molecule-1 (VCAM-1) and interferon-gamma-induced protein (IP-10) in HIV-infected patients. To delineate the mechanisms underlying this therapeutic effect, we tested whether clinically relevant concentrations of PTX suppress VCAM-1 or IP-10 release in cultivated human lung microvascular endothelial cells. Indeed, we found that tumor necrosis factor (TNF)-α-induced VCAM-1 was reduced with concentrations of PTX in the low nanomolar range, comparable to plasma levels in PTX-treated groups. We also investigated the effect of HIV proteins and found that HIV transactivator of transcription (HIV-Tat) and HIV-envelope-derived recombinant gp120 enhanced TNF-α-induced VCAM-1 gene expression in lung microvascular and coronary macrovascular endothelial cells, respectively. In addition, PTX and a NF-κB-specific inhibitor reduced this enhanced VCAM-1 gene induction in microvascular and macrovascular endothelial cells. These results provide novel insights in how the antiinflammatory agent PTX can directly reduce HIV-associated proinflammatory endothelial activation, which may underlie vascular dysfunction and coronary vascular diseases.
Introduction
C
It is widely accepted that endothelial activation and dysfunction are involved in the early development of atherosclerotic disease. Proinflammatory cytokines, including tumor necrosis factor-α (TNF-α) and interferon-γ (IFN-γ), can induce both endothelial dysfunction and atherosclerosis. 9,10 Whereas the effects of TNF-α in endothelial cells are primarily mediated through NF-κB and secondarily through the p38 MAPK pathways, 11 –13 the effects of IFN-γ are signaled primarily through the JAK-STAT pathways. 14 However, little is known about whether HIV viral proteins can directly contribute to atherosclerosis. The HIV envelope protein gp120 and the HIV transactivating protein Tat, which are secreted from HIV-infected cells, can activate endothelial cells. 15,16 Therefore, it is likely that HIV infection can cause endothelial activation and dysfunction by both direct (Tat and gp120) and indirect (release of cytokines from mononuclear cells) means, thus promoting atherogenesis or atherogenic plaque progression.
To assess the endothelial mechanism of early atherogenic events in HIV-infected patients, flow-mediated dilation (FMD) as an in vivo marker for endothelial function was employed. In these studies, salsalate (a salicylic acid-generating drug) and pentoxifylline (PTX) were demonstrated to improve endothelial function in HIV-infected persons not receiving cART. 17 –19 While salsalate treatment was based on the initially description of its correlate sodium salicylate as an NF-κB signal transduction inhibitor, 20 PTX was originally discovered as a PDE-1 inhibitor and later demonstrated to also inhibit NF-κB. 21 –24 In addition to improving FMD values, treatment with PTX reduced soluble levels of vascular cell adhesion molecule-1 (VCAM-1) and interferon-gamma-induced protein (IP-10) after 8 weeks of treatment. 18 Soluble VCAM-1 is a surrogate marker for the transmembrane form of the endothelial adhesion molecule VCAM-1, which mediates leukocyte adhesion and is linked to endothelial dysfunction and to the early onset of atherosclerosis. 25 The chemokine IP-10 has been previously described to promote two key processes involved in atherosclerosis, namely Th1 immune system activation and endothelial apoptosis. 26,27
In the present study, we tried to determine whether we could identify the cellular mechanisms underlying the beneficial effects observed in our clinical PTX treatment trial. Thus, we investigated the role of the HIV-1 proteins gp120 and Tat in combination with proinflammatory cytokines in the presence and absence of PTX in both primary human lung microvascular endothelial cells (HLMVECs) and human coronary arterial endothelial cells (HCAECs). We chose the first cell type because the lung has the highest number of endothelial cells of all organs and is thus likely to have contributed greatly to the observed reduction of sVCAM-1 and IP-10 levels in our PTX trial. 17,18 The second endothelial cell type was used because of its physiological relevance to HIV-associated cardiovascular disease. Our in vitro data demonstrate that HIV proteins can enhance TNF-α-induced endothelial cell activation as measured by induction of VCAM-1 and that PTX can reduce this proinflammatory activation.
Materials and Methods
Reagents
Recombinant Tat and gp120 proteins were purchased from ImmunoDiagnostics (Woburn, MA). VCAM-1-PE antibody was purchased from Abcam. IKK inhibitor SC-514 was purchased from Calbiochem (La Jolla, CA). All other reagents including PTX and cytokines were obtained from Sigma (St. Louis, MO).
Cell culture
HLMVECs and HCAECs were obtained from Lonza (Allendale, NJ) and maintained in culture medium consisting of EMB-2, 5% FBS, 0.4% hydrocortisone, 1.6% hFGF, 1% VEGF, 1% IGF-1, 1% ascorbic acid, 1% hEGF, 1% GA-100, and 1% heparin. All primary cell cultures were maintained at 37°C in 5% CO2 and 95% air. Culture medium was changed every 2 days.
In vitro assays
For analysis of VCAM-1 and IP-10 gene expression and protein release, 4.0×104 HLMVECs between passages 3 and 6 were seeded in each well of 12-well tissue culture plates. After 2 days culture medium was changed, and after a further 17 h reagents were added without changing the medium again. After the indicated time points, the supernatant was harvested for IP-10 analysis and the cells were either removed from the plate using 5 mM EDTA in phosphate-buffered saline (PBS) for staining analysis or lysed and the RNA harvested using a NucleoSpin RNA isolation kit (Clontech Laboratories, Mountain View, CA) for reverse transcriptase polymerase chain reaction (RT-PCR).
Real-time qRT-PCR analysis
Total RNA was extracted and reactions were performed in duplicate on 96-well optical reaction plates with one-step SYBR Green PCR master mix (iScript One-Step RT-PCT Kit with SYBR Green, Bio-Rad Laboratories, Hercules, CA). The real-time PCR reaction and expression quantification was performed in a Chromo4 Opticon analyzer instrument (Bio-Rad, Hercules, CA). Then 50 ng of total RNA was added to each well and the thermal cycling conditions were as follows: 50°C for 10 min (cDNA synthesis), 95°C for 5 min (reverse transcriptase inactivation), followed by 42 cycles of 95°C for 10 s and 58°C for 30 s. Primers used for the RT-PCR include CTCCAGTCTCAGCACCATGA (FW) and CAAAATTGGCTTGCAGGAAT (RE) for IP-10 as well as TGTTGAGATCTCCCCTGGAC (FW) and GAATTGGTCCCCTCACTCCT (RE) for VCAM-1. To compare the levels of gene expression between the experimental groups, a second derivative calibration method for relative quantification was employed as described previously. 12 The amount of the target gene transcript was normalized to eF1α (elongation factor alpha), and measured relative to a calibrator. The target gene expression was computed by the 2 – ΔΔC t method, where C t is the crossing point or turning point of the fluorescence curve calculated by analyzer instrument software, ΔC t of target or calibrator is the C t of the target gene subtracted from the C t of the housekeeping gene, and ΔΔC t is ΔC t (unknown target gene) – ΔC t (calibrator).
Immunofluorescence and MetaMorph analysis
For immunofluorescence of surface VCAM-1 expression, HLMVECs were plated at 4.0×104 cells per well of a four-well chambered slide (Nunc) and cultured for 2 days. Cells were treated with or without different factors and incubated for 6 h then washed three times with wash buffer [0.5% bovine serum albumen (BSA), 0.05% Tween 20 in PBS], and nonspecific antigens were blocked with universal blocking solution (Dako, Carpenteria, CA). The samples were incubated with a 1:100 dilution of rabbit antihuman VCAM-1 antibody (clone H-276; Santa Cruz Biotechnology, Santa Cruz, CA) in a moist chamber for 16 h at 4°C, followed by three washes in PBS and incubation with 1:200-diluted Alexa Fluor 488-conjugated goat antirabbit IgG (Invitrogen, Carlsbad, CA) for 1 h in a moist chamber at 37°C. Cells were then treated with 10 μg/ml DAPI for 5 min and the cells were mounted under CC-mount (Sigma, S. Louis, MO) and analyzed on a Nikon Eclipse (TE200S) inverted fluorescence microscope. For the negative control reaction some wells were either incubated with nonimmune isotype-matched control IgG or omitted primary antibody. About six images per well were captured with a 20×objective using Voxx image software in a blinded fashion and quantitative intensity (expression) data were obtained by MetaMorph Imaging software (Molecular Devices, Sunnyvale, CA). Values for VCAM-1 staining were normalized to the number of nuclei for every image in both treated and untreated groups. Data were presented as mean±SEM of each treatment normalized to unstimulated cells performed in triplicate and repeated independently two additional times.
Flow cytometry
After seeding of HLMVECs or HCAECs, treatment, and subsequent harvesting as described above, cells were incubated with a 1:10 dilution of anti-VCAM-1-PE (Abcam) for 1 h at 4°C then washed three times with PBS and fixed with 4% PFA for 5 min at RT. FACS analysis was performed using a FACS Calibur II (Beckton Dickenson) and analyzed using CellQuest software.
Statistical analysis
Comparisons were made using both two-tailed and paired Student's t-tests as appropriate. A p-value <0.05 was considered significant.
Results
Pentoxifylline reduces TNF-α-induced VCAM-1 at low concentrations consistent with those observed in the serum of treated subjects
We questioned whether PTX could reduce TNF-α-induced VCAM-1 or IFN-γ-induced IP-10 gene activation in human HLMVECs at concentrations found in individuals treated with PTX (median plasma PTX concentrations were between 270 and 340 nM). 19 For TNF-α (20 ng/ml) and IFN-γ (40 ng/ml), we employed concentrations previously used to stimulate endothelial cells. 12,28 Pentoxifylline substantially inhibited the expression level of TNF-α-induced VCAM-1 in a dose-dependent manner starting at a concentration as low as 8 nM (Fig. 1A). In contrast, IFN-γ-induced expression of the chemokine IP-10 was reduced by PTX only at much higher concentrations (50 and 250 μM) than those achieved in PTX-treated patients (Fig. 1B). Therefore, we focused on the role of TNF-α-induced VCAM-1 production in the following experiments.
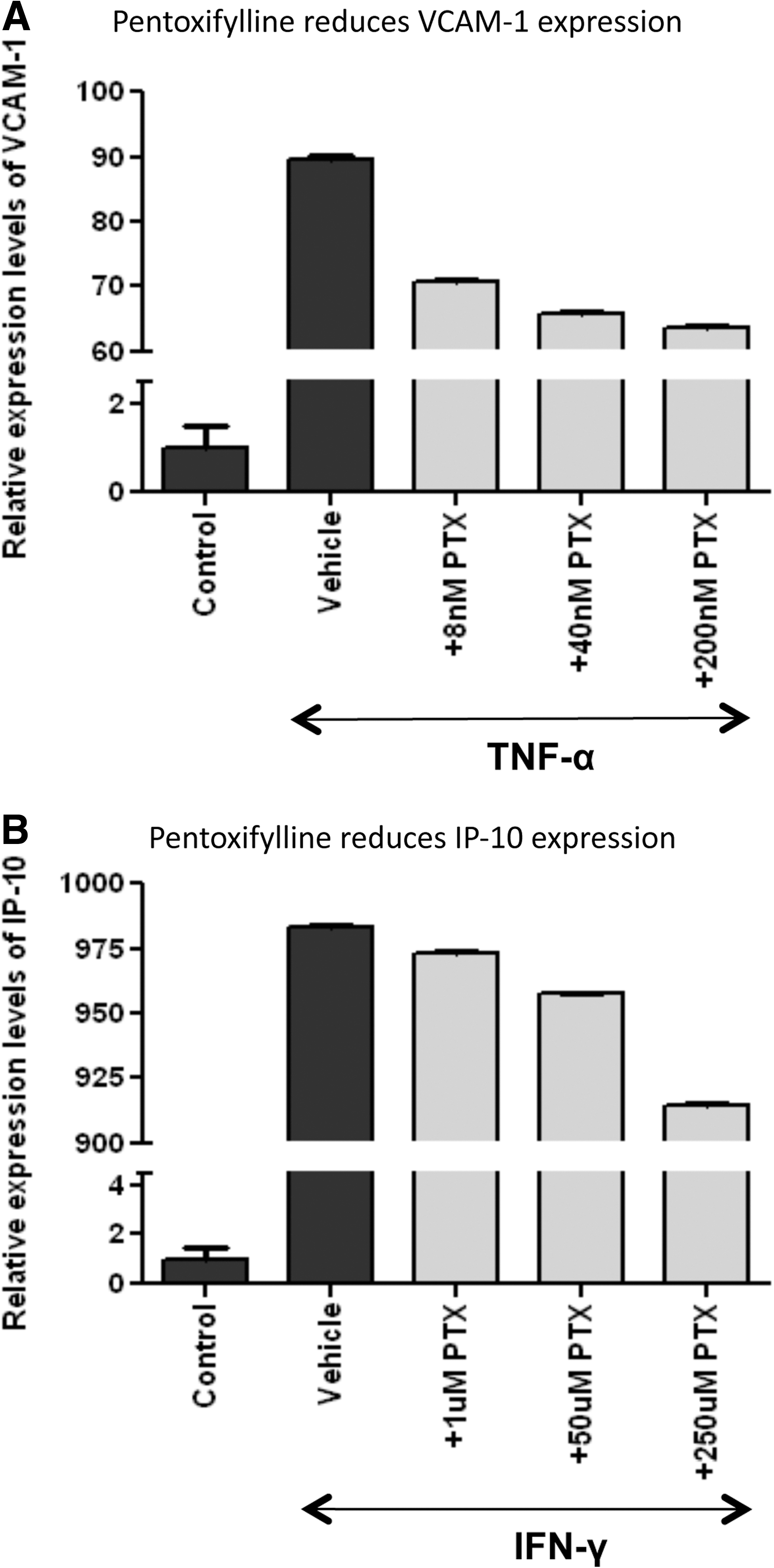
Pentoxifylline reduces tumor necrosis factor (TNF)-α-induced vascular cell adhesion molecule-1 (VCAM-1) at low concentrations. Human lung microvascular endothelial cells (HLMVECs) were incubated for 6 h with either
HIV transactivator of transcription (HIV-Tat) enhances TNF-α-induced VCAM-1 expression in HLMVECs
To explain the effects of reduced sVCAM-1 levels in serum of PTX-treated HIV-infected individuals, we first tested whether secreted HIV proteins can enhance TNF-α-induced VCAM-1 production and whether this induction was reversible with PTX. We cotreated cells with HIV-Tat
29
and TNF-α to mimic the inflammatory state found in patients with HIV.
30
–32
As shown in Fig. 2A, exposure of HLMVECs to recombinant Tat significantly increased the gene expression level of VCAM-1 in combination with TNF-α. Importantly, while we used the concentration of Tat resulting in the highest increase (100 ng/ml), a dose dependency revealed significantly more VCAM-1 induction at a lower concentration of 50 ng/ml Tat in the presence of TNF-α (Supplementary Fig. S1A; Supplementary Data are available online at
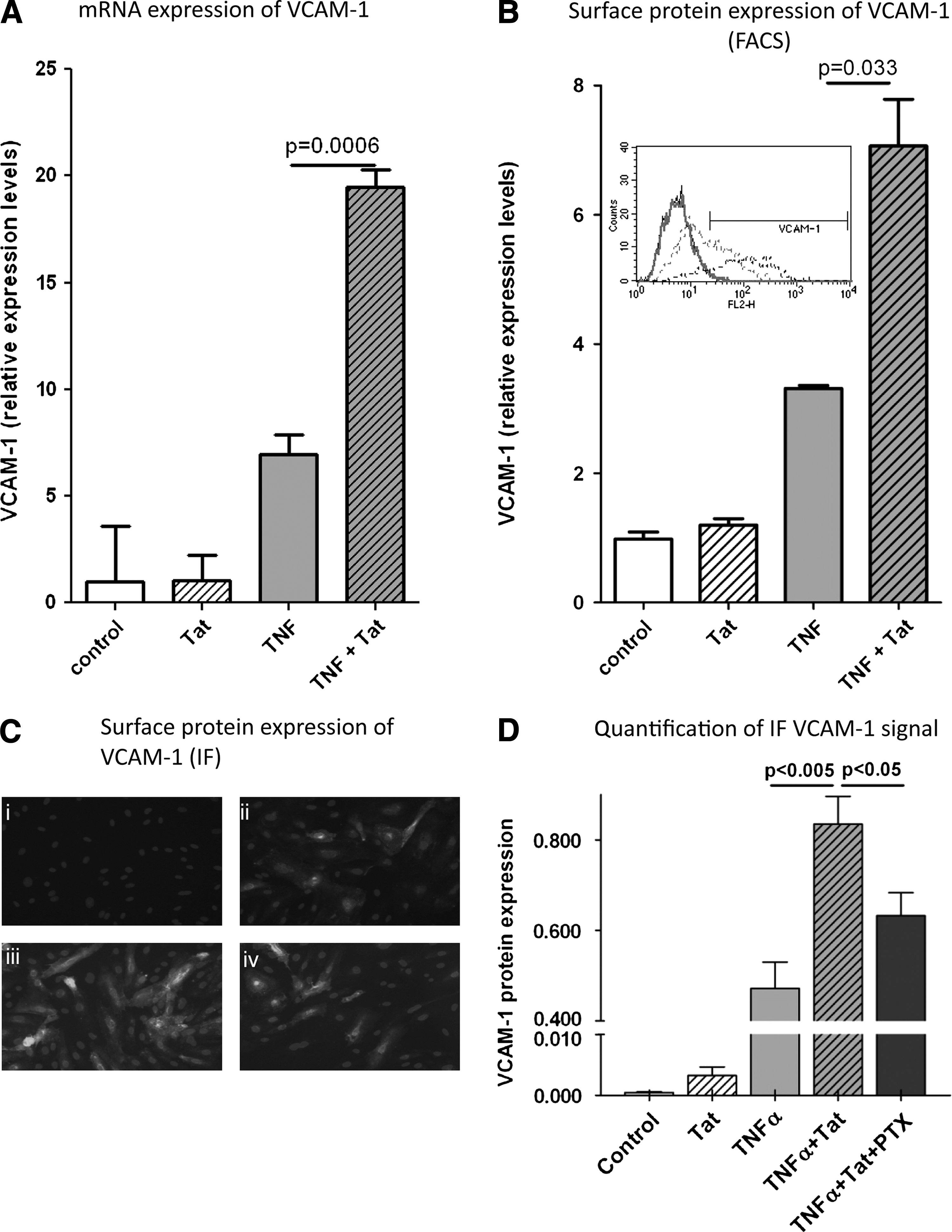
HIV transactivator of transcription (HIV-Tat) enhances TNF-α-induced VCAM-1 expression in HLMVECs. HLMVECs were treated with low TNF-α concentrations (1 ng/ml) alone or in combination with HIV-Tat (100 ng/ml) for 6 h.
gp120 enhances TNF-α-induced VCAM-1 expression in HCAECs
To confirm the pathophysiological significance of our findings in human lung microvascular endothelial cells, we extended our studies to HCAECs. While Tat did not significantly increase TNF-α-induced VCAM-1 production in HCAECs (data not shown), gp120 strongly enhanced TNF-α-induced VCAM-1 production in HCAECs at both mRNA (Fig. 3A) and protein (Fig. 3B) levels. Once again, we used the maximal concentration of gp120 (100 ng/ml) although gp120 in a comparable dose dependency as observed with Tat already showed a robust enhancement of TNF-α-induced VCAM-1 expression at 50 ng/ml (Supplementary Fig. S1B).
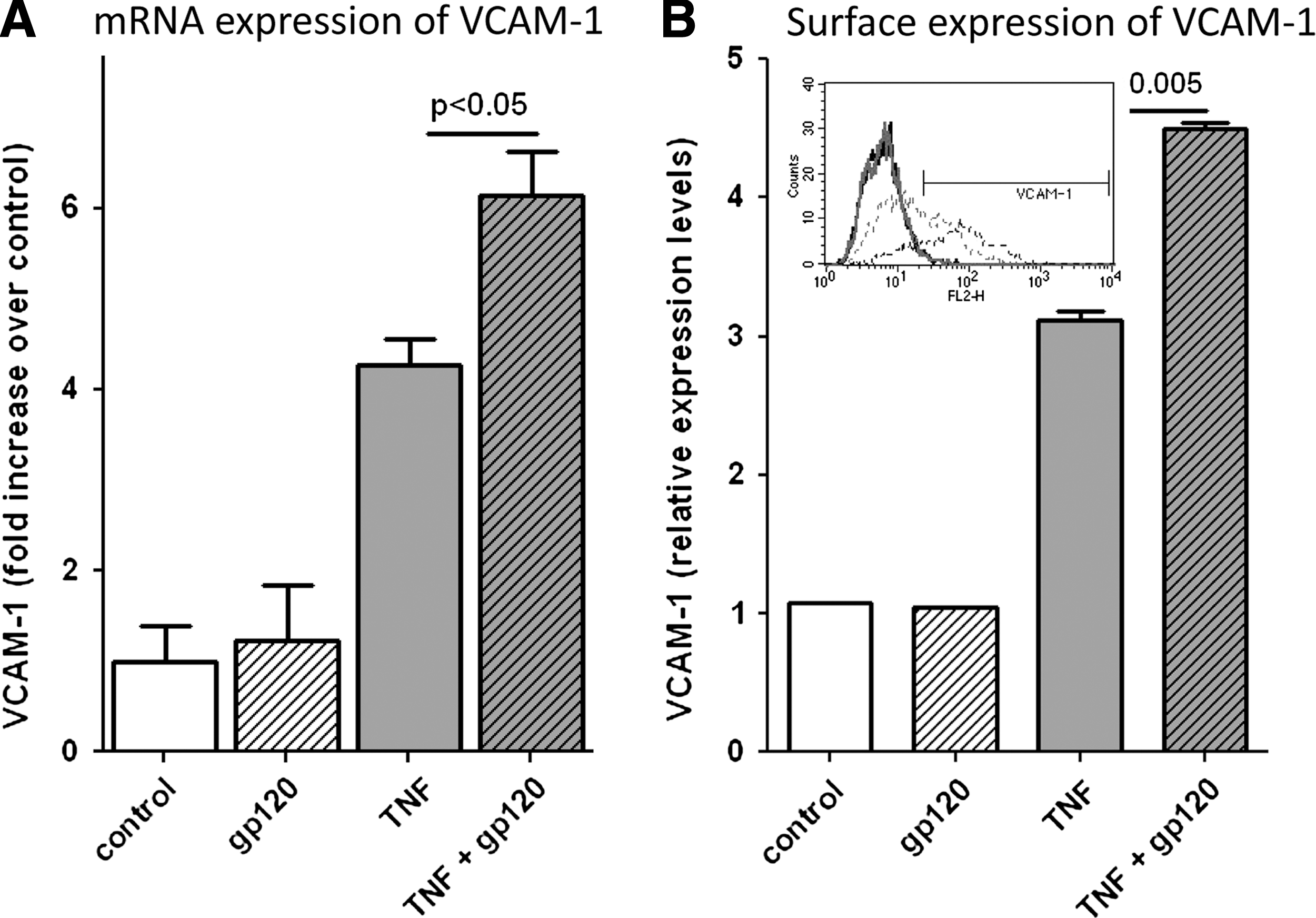
gp120 enhances TNF-α-induced VCAM-1 expression in HCAECs. HCAECs were treated with TNF-α (1 ng/ml) alone or in combination with gp120 (100 ng/ml) for 6 h and then analyzed for
Pentoxifylline and IKK inhibition reduce Tat- and gp120-induced upregulation of VCAM-1
TNF-α signals primarily through the NF-κB pathway, 33 which is also inhibited by PTX. Therefore, we compared the effect of an IKK inhibitor with PTX on the upregulation of VCAM-1. HMLVECs and HCAECs were treated with TNF-α and Tat or gp120, respectively, in the absence and presence of 200 nM PTX or 100 nM of the IKK inhibitor SC-514 (IKKI), two concentrations that we had established to maximally reduce TNF-α-induced VCAM expression (Fig. 1A and data not shown). After 6 h cells were either harvested for RNA, with the levels of VCAM-1 RNA determined by RT-PCR (Fig. 4A and C), or were stained for surface VCAM-1 and analyzed by flow cytometry (Fig. 4B and D). In both cell types the RNA levels and surface VCAM-1 decreased upon treatment with PTX or the IKK inhibitor, although the effect on mRNA was much more pronounced with the IKK inhibitor. In contrast, surface VCAM-1 reduction was similar upon treatment with PTX or IKK inhibitor; the reductions were not as dramatic as with mRNA levels. Although the time course of VCAM-1 expression in endothelial cells is known to be between 4 and 12 h, we also repeated these experiments with 12 h of treatment and had very similar results (data not shown). We further confirmed the effect of PTX on VCAM-1 expression using a lower (50 ng/ml) concentration of Tat and gp120 (Supplementary Fig. S2A and C). However, the higher Tat and gp120 concentrations displayed borderline or no significant synergies with TNF-α, respectively, and inhibition with PTX was not significant any more (Supplementary Fig. S2B and D).
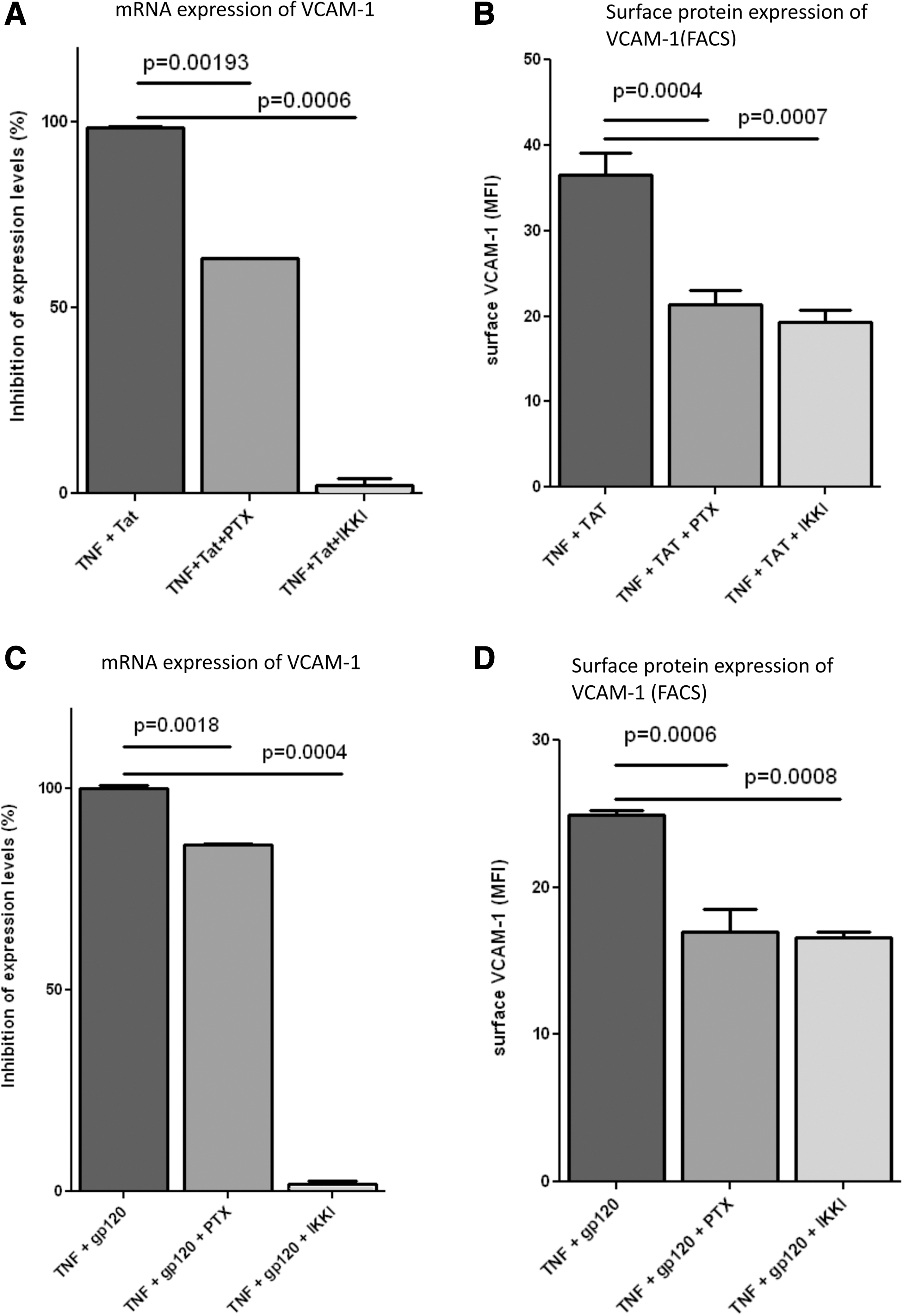
Tat- and gp120-enhanced TNF-α-induced upregulation of VCAM-1 displays differential sensitivity to PTX and IKK inhibitors. HLMVECs were treated with TNF-α (1 ng/ml) in combination with HIV-Tat (100 ng/ml) with/without PTX (200 nM) or IKKI (100 nM) for 6 h and then analyzed for
Discussion
This study shows for the first time that secreted HIV proteins strongly augment endothelial production of the proinflammatory adhesion molecule VCAM-1 in microvascular and arterial macrovascular endothelial cells when combined with the inflammatory cytokine TNF-α. These in vitro results suggest that HIV proteins, in concert with the proinflammatory cytokines found in HIV-infected patients, can synergistically increase endothelial inflammation in adult coronary and microvascular endothelial cells, similar to what has been previously shown for the HIV protein Tat in umbilical cord endothelial cells. 29 Surprisingly, we found Tat enhanced TNF-α-induced VCAM-1 induction only in human lung microvascular endothelial cells, whereas gp120 enhanced TNF-α only in human coronary arterial endothelial cells. In all cases the observed effects were significantly reduced upon treatment with the NF-κB inhibitors SC-514 and PTX. 21 –24,34,35 This response seems to be specific for TNF-α, as we also tested the combination of IFN-γ and Tat with and without PTX. These results were negative in two ways: first, there was no significant additional upregulation of VCAM-1 by the combination of Tat and IFN-γ, and second, PTX treatment did not result in lower VCAM-1 expression as compared to IFN-γ alone (data not shown). Together, these findings highlight the direct link between HIV infection, inflammation, and vascular dysfunction on the level of the endothelium and emphasize the significance of our previous finding that PTX can reduce vascular dysfunction in the endothelium. 19
To address the mechanism of reduction of soluble VCAM-1 levels in PTX-treated HIV-infected patients not on antiretroviral therapy, we have tested for VCAM-1 induction in human lung microvascular endothelial cells, as these are the most common type of endothelial cell in humans. Our data can explain the PTX-dependent reduction of sVCAM-1, which is a surrogate marker for endothelial VCAM-1 production, in HIV-positive individuals. 36,37 To test our hypothesis that cooperation between HIV and inflammatory cytokines may be a mechanism leading to HIV-associated cardiovascular disease, we assessed for a potential HIV and TNF-α synergy in HCAECs. In this study we focused on the effect of HIV proteins on VCAM-1 since it was upregulated in the in vivo trials and is specific for endothelial cells, whereas other TNF-regulated inflammatory proteins, such as ICAM-1 and IL-6, are also expressed in other cell types. Although we do not anticipate different regulation with the majority of these genes based on our finding that PTX predominately inhibits the NF-κB pathway, we cannot verify this with all proteins. Indeed, our preliminary data show that ICAM-1 exhibits the same enhanced expression with HIV proteins as VCAM-1 (data not shown). Further studies into the specifics of TNF-regulated inflammatory proteins and the differences between endothelial expression and that of other cell types are needed.
Surprisingly, Tat did not significantly enhance TNF-α-induced VCAM-1 expression in HCAECs under conditions we applied for microvascular endothelial cells. However, gp120 strongly enhanced TNF-α-induced VCAM-1 expression in HCAECs, which is in line with a previous study demonstrating that HIV gp120 and TNF-α synergistically reduce eNOS expression. 38 Interestingly, gp120 had no effect by itself or on the TNF-α-induced VCAM-1 expression in microvascular HLMVECs.
TNF-α signals primarily through the NF-κB pathway 33,39 and PTX inhibits NF-κB signal transduction. 22,23 Therefore, we addressed the role of NF-κB for enhanced TNF-α-induced VCAM-1 expression in HCAECs using a more specific IKK inhibitor. The IKK inhibitor was stronger than PTX when it came to blocking VCAM-1 upregulation on gene expression, whereas surface expression levels were similar in PTX- and IKKI-treated cells. This could be explained by the possibility that surface expression also is dependent on several regulatory steps including protein translation, trafficking, and protein stability. Alternatively, this discrepancy could be explained by differential kinetics between TNF-α-induced VCAM-1 upregulation and PTX or IKKI inhibition of mRNA. If VCAM-1 upregulation occurs prior to the effects of PTX or IKKI, there would still be a small amount of VCAM-1 expressed on the surface before inhibition of transcription, and this amount would be consistent between all samples treated with TNF-α and Tat or gp120.
Upon treatment with PTX, VCAM-1 inhibition occurred at concentrations as low as 8 nM, whereas IP-10 inhibition required more than 25,000-fold higher concentrations, at ∼250 μM (Fig. 1). One possible reason for the difference in sensitivity to PTX in TNF-α- and IFN-γ-treated cells is that IFN-γ signals through the JAK–STAT pathway rather than the NF-κB signaling pathway. 14 Although we did not observe a reduction of IP-10 production at physiologically relevant concentrations of PTX here, IP-10 was significantly reduced in patients. 19 This discrepancy may be because we tested only endothelial cells in this study and, unlike VCAM-1, IP-10 is produced by multiple cell types; therefore, the in vivo reduction of IP-10 levels may be due to a reduction in other cell types. Of note, our dose-response curve in human lung microvascular endothelial cells shows PTX to work at a much lower dose than reported in human umbilical cord vein endothelial cells. 40,41 Again, it could be a property of lung microvascular endothelial cells to metabolize PTX differently than other endothelial cells. Finally, the PTX-dependent inhibition of VCAM-1 expression was incomplete, suggesting that there is at least one other pathway in addition to NF-κB-dependent signaling that may contribute to the upregulation of VCAM-1. In this context the previously described ability of the HIV-Tat protein to induce oxidative and inflammatory pathways in the endothelium is an important finding. 16 One potential pathway could involve MAP kinases, which have been shown to be activated by HIV-Tat. 42,43 These are significant because perturbation of MAP kinase signaling has been shown to be significant in HIV infection; for example, increased signaling can increase LPS-mediated HIV-1 transcytosis across the blood–brain barrier, 44 a possible key step in the development of HIV-associated dementia.
VCAM-1 has been identified as a novel prognostic marker for cardiovascular diseases 8,45,46 and is suggested to promote processes of atherosclerosis by recruiting monocytes and T cells to the sites of the beginning plaques. 25 It may be that infected monocytes and T cells then further contribute to vascular dysfunction by increasing the local concentration of Tat and gp120. Inflammation is a prerequisite because endothelial activation by Tat or gp120 alone was low in comparison to that induced by TNF-α (Fig. 3). Importantly, very high concentrations of Tat and gp120 showed reduced synergy with TNF-α and inhibition with PTX (Supplementary Fig. S2A and C), indicating that in extremely high viremia PTX could be less protective against HIV-1-induced endothelial dysfunction. Since antiretroviral therapy in the absence of antiinflammatory therapy only partially restores endothelial function, 47 adding PTX may provide additive benefits by controlling residual inflammation and may thus further improve endothelial function in patients receiving antiretroviral therapy. These findings also have implications for the treatment of lung abnormalities as well as cardiovascular disease in the HIV-positive population. HIV-infected patients exhibit increased incidence of lung disorders such as pulmonary edema, ARDS, and emphysema. 48 –51 Interestingly, the development of ARDS can be predicted by the levels of sVCAM-1 in the serum, much like cardiovascular disease. 52 Therefore, it is likely that treatment with PTX will not only decrease the severity of cardiovascular risk in HIV patients but may also reduce the risk of pulmonary disorders.
Together, our in vitro data in combination with our in vivo clinical studies can lead to a more complete mechanistic understanding of the links among HIV infection, inflammation, and endothelial activation and dysfunction. These results may subsequently lead to the development of novel antiinflammatory approaches to reduce cardiovascular events in HIV-infected patients.
Footnotes
Acknowledgments
This research was supported by grants from the National Institutes of Health (R01 HL095149) and the Indiana University Research Support Funds Program.
Author Disclosure Statement
No competing financial interests exist.