
Abstract
Chronic HIV-1 infection results in the expansion of both NKG2C+ and CD16+CD56− human natural killer cells. NKG2C+ cells proliferate in response to human cytomegalovirus (HCMV) and expansion of the dysfunctional CD56−CD16+ natural killer (NK) cells is associated with HIV-1 viremia. Here we report an association between increased proportions of CD56−CD16+ NK cells in viremic HIV-1+ individuals and an increased contribution of NKG2C+ cells to this subset. These data, in addition to anti-HCMV IgG serology, indicate a potential contribution of both HCMV and HIV-1 to NK cell dysfunction in HIV-1-infected individuals.
N
The major population of NK cells defined by the expression of both CD56 and CD16 is diminished in the blood both in absolute number and as a proportion of total NK cells. 4,5 Parallel to such NK cell losses, a dysfunctional NK cell population emerges, characterized by a loss of CD56, while the expression of CD16 is maintained. 4 –6 Loss of function in these CD56−CD16+ NK cells is attributed to elevated expression of inhibitory killer cell immunoglobulin-like receptors (KIR) and loss of activating natural cytotoxicity receptors (NCR), both of which impact NK cell–dendritic cell interactions. 5,7 HIV-1+ individuals with a high plasma viral load (greater than 10,000) have a higher proportion of CD56−CD16+ NK cells compared to those with lower viral loads, irrespective of treatment with antiretroviral therapy, or aviremic individuals receiving highly active antiretroviral therapy (HAART), indicating a direct role for HIV-1 viremia in the generation of dysfunctional NK cells. 8
We and others have also demonstrated that an NK cell subset defined by a c-type lectin like receptor NKG2C, which is involved in the recognition of human cytomegalovirus (HCMV), is also expanded in individuals chronically infected with HIV-1 compared to HIV-1-negative individuals. 9 –11 NKG2C+ NK cells are absent or are present only at low frequency in individuals seronegative for antibody against HCMV (HCMV-IgG), indicating a dominant role of HCMV in the emergence and expansion of this NK cell subset. 9,11,12 In common with “dysfunctional” CD56−CD16+ NK cells, the NKG2C+ NK cell subset also expresses low NCR and elevated KIR expression. 10,12 Furthermore, the ratio of NKG2A+ to NKG2C+ NK cells is inverted in CMV-seropositive individuals with chronic HIV-1 viremia. 13 However, the proportions of NKG2C+ NK cells in these studies were calculated only after gating on CD56+ NK cells and so it was therefore not possible to assess whether these cells made a significant contribution to the “dysfunctional” CD56−CD16+ NK cell subset in HIV-1-infected individuals. We hypothesized that NKG2C and CD56−CD16+ NK cell subsets may be overlapping with the implication that HCMV, in addition to HIV-1 viremia, could be responsible for driving the appearance of the dysfunctional CD56−CD16+ cell subset in HIV-1+ individuals. To test this hypothesis we performed multiparameter flow cytometric analysis to investigate the expression of NKG2C on different NK cell subsets defined according to CD56 and CD16 expression.
Blood was taken with full ethical permission and informed consent (Nation Research Ethics Committee, UK and Riverside Research Ethics Committee, London, UK). NK cell subsets were compared in HIV-1+ aviremic individuals who had received HAART without detectable virus for 12 months prior to the study (n=80) and HIV-1+ individuals with low plasma viral load (n=32) and in those with a plasma viral load of 10,000 or greater at the time of the study (n=23). HIV-1-negative control individuals were also studied (n=32). Characteristics for patient and control groups are shown in Table 1.
Peripheral blood mononuclear cells were labeled with the following monoclonal antibodies: anti-CD3, anti-CD14, and anti-CD19 PerCP (Becton Dickinson, Oxford UK), anti-CD56 APC (Beckman Coulter, Marseille, France), anti-CD16 FITC (Serotec, Abingdon, UK), and anti-NKG2C phycoerythrin conjugate (R&D Systems, UK). CD56−CD16+, CD56+CD16+, and CD56+CD16− NK cell subsets were identified within lymphocytes after exclusion gating on CD3+, CD14+, and CD19+ cells and CD56−CD16− cells (see Fig. 1). The proportions of total NK cells and each NK cell subset coexpressing NKG2C were then calculated.
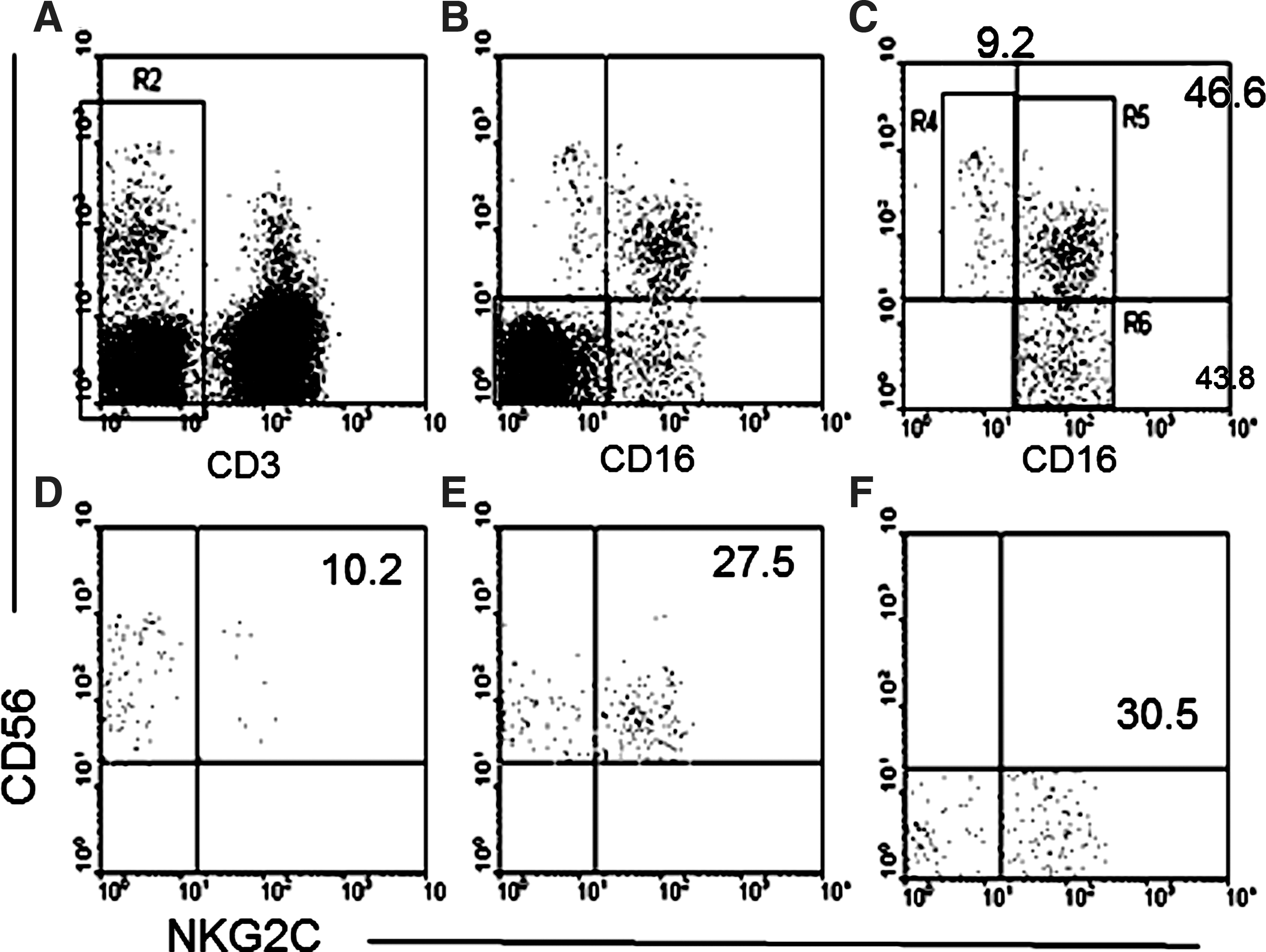
Gating strategy for detection of NKG2C+ natural killer (NK) cells within CD56+CD16−, CD56+CD16+, and CD56−CD16+ NK cell subsets.
Titers of anti-HCMV-IgG were determined using a commercially available ELSIA kit (BioKit, Spain). Anti-HCMV-IgG was detectable in 16 out of 32 (50%) HIV-1-seronegative control individuals, in 76 out of 80 (95%) aviremic HIV-1+ individuals, and in 100% of viremic HIV-1+ individuals.
Statistical analyses were performed using StatView 5.01 software. The Mann–Whitney U test for comparison between groups and linear regression analysis was performed using Spearman-Rank correlation. p values of less than 0.05 were considered significant.
To assess the contribution of NKG2C+ NK cells to the dysfunctional CD56−CD16+ NK cell subset we performed a cytometric gating strategy to exclude non-NK cells and to include CD56+ and CD56− NK cell subsets in our analysis. Figure 1 shows a representative analysis of NK cells from an HIV-1+ individual with high plasma viral load. T cells (CD3+), monocytes (CD14+), and B cells (CD19+) and subsequently remaining CD16−CD56− cells were first excluded (Fig. 1, upper left and central panels) and NK cell subsets were subsequently gated into CD56+CD16−, CD56+CD16+, and CD56−CD16+ populations (Fig. 1, upper right for analysis of NKG2C expression; lower panels). Initial analysis of the proportions of CD56+CD16−, CD56+CD16+, and CD56−CD16+ subsets within total NK cells demonstrates that significantly higher proportions of CD56−CD16+ NK cells are found in both aviremic (median 14.2%, range, 1.7–54.5%) and viremic HIV-1+ patients (low VL, median 16.8%, range 6.7–64.1%; high VL, median 19.3%, range 4.4–74.5%) compared to HIV-1-negative control individuals (median 7.2%, range 1.2–23.7%, p<0.0001 for all comparisons). Furthermore, HIV-1+ individuals with high plasma viral load have a significantly higher proportion of CD56−CD16+ NK cells compared to aviremic HIV-1-infected patients (p=0.0359). The proportion of CD56+CD16+ cells was correspondingly significantly higher in HIV-1-negative individuals (median, 87.1%, range, 52.6–95.5%) compared to aviremic (median, 72.6%, range, 15.3–93%, p<0.0001) and viremic HIV-1+ individuals (low VL, median 69.1%, range 13.5–92%, p<0.0001; high VL, median 70.6%, range 1.4–90.4%, p=0.0003) (Fig. 2A).

The proportion of total NK cells of CD56−CD16+, CD56+CD16+, and CD56+CD16+ phenotype
The overall proportion of NKG2C+ cells detected within all NK cells was significantly elevated in aviremic (median 27.3%, range 1.0–77.0%, p<0.0001) and viremic HIV-1-infected patients (low VL, median 17.9%, range 0.6–74.8%, p<0.0001; high VL, median 36.1%, range 4.3–94.0%, p<0.0001) compared to HIV-1-negative control individuals (median 5.8%, range 2.4–30.2%) (Fig. 2B). In contrast to CD56−CD16+ NK cells, control of HIV-1 plasma viral load by antiretroviral therapy had no effect on the proportion of NKG2C+ NK cells, as previously reported. 10 No relationship was observed between CD4+ T cell count and the proportions of CD56−CD16+ or NKG2C+ NK cells. We then assessed the proportions of NKG2C+ cells within gated NK cell functional subsets to see if NKG2C+ cells contributed to the CD16+ CD56− subset and if this receptor was enriched within any particular NK subset (Fig. 2C).
As expected, fewer NKG2C+ NK cells were observed in any NK cell subset from HIV-1-seronegative healthy control individuals, including CD56+CD16+ cells (median, 6.4%, range, 1.1–72.7%), whereas these were predominantly observed within the CD56+CD16+ NK cell subset from aviremic (median, 30.5%, range 0.1–90.1%, p<0.0001) and viremic (low VL, median 24.4%, range 0.1–86.5%, p<0.0001; high VL, median 27.3%, range 5.6–94.9%, p<0.0002) HIV-1+ individuals with a significant increase above the level observed in HIV-1-seronegative controls (Fig. 2C). NKG2C expression was detected on CD56−CD16+ NK cells from HIV-1-infected individuals and was significantly elevated within this subset in HIV-1-infected individuals with high viral load (median, 24.8%, range 2.9–82.6%) compared to HIV-1-seronegative control individuals (median, 11.3%, range 3.9–54.7%, p=0.037) (Fig. 2C).
Interestingly, the expression of NKG2C was elevated within CD56+CD16− NK cells from aviremic HIV-1+ individuals (median, 23.2%, range, 0.1–60.0%) compared to viremic individuals (low VL, median, 13.5%, range, 0.1–50.0%, p=0.032, high VL, median, 14.6, range, 0.1–62.7%, p=0.013) indicating a potential shift toward expression on immature NK cells with antiretroviral therapy.
In view of the differential effect of HIV-1 plasma viremia on CD56−CD16+ NK cells and the enrichment for NKG2C+ cells within CD56−CD16+ NK cells from HIV-1-infected individuals with high plasma viral load, the relationship between these subsets was assessed by correlating the overall proportion of CD16+CD56− NK cells with the contribution of NKG2C+ NK cells to this subset. A strong correlation was observed between the overall proportion of CD56−CD16+ NK cells and the percentage of these cells expressing NKG2C in individuals with a high HIV-1 plasma viral load (Fig. 3B, r=0.836, p<0.0001), whereas this was not observed in HIV-1-seronegative controls (Fig. 3A, r=0.103, p=0.581). No correlation was observed between the expression of NKG2C and the proportion of CD56−CD16+ cells in individuals with low or undetectable plasma viral load (r=0.026, p=0.884 and r=0.064, p=0.561, respectively, data not shown).
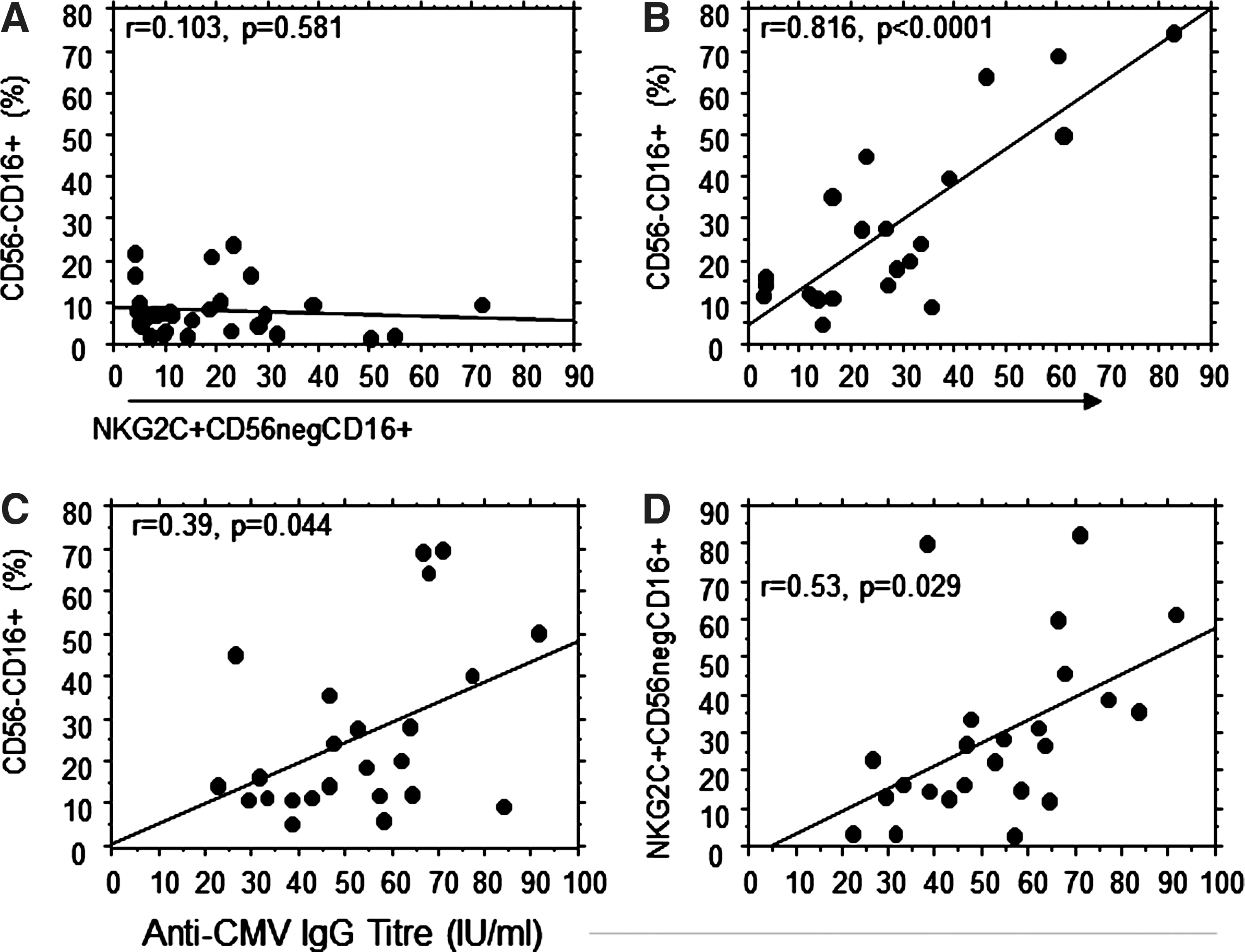
The proportion of CD56−CD16+ NK cells correlates directly with the proportion of NKG2C+ cells within this subset in HIV-1-seronegative controls
A significant elevation in the titer of plasma IgG recognizing pp65 of HCMV was detected in all three groups of HIV-1+ individuals compared to HIV-1-seronegative controls, indicative of greater exposure to HCMV (Fig. 2D). The relationship between HCMV antibody titer and the proportion of total CD56−CD16+ and NKG2C+ CD56−CD16+ NK cells was also assessed to test whether exposure to HCMV could contribute to the increase in this subset in HIV-1+ individuals. A weak correlation was observed between the titer of anti-HCMV IgG only in individuals with high HIV-1 viremia (total CD56−CD16+, r=0.39, p=0.044; NKG2C+ CD56−CD16+, r=0.513, p=0.029), indicating a potential role for HCMV exposure in promoting these NK cell populations (Fig. 3C and D).
These data demonstrate that NKG2C+ NK cells preferentially acquire a CD56− phenotype in individuals with high HIV-1 plasma viral load. Changes to the NK cell repertoire in HIV-1-infected individuals, including a loss of NCR and an increased inhibitory KIR expression, may largely be directed by major concomitant infections such as HCMV. Progression of these NKG2C+KIR+NCRlow/− cells toward a dysfunctional CD56− phenotype may subsequently be driven either by HIV-1 itself or by other factors associated with chronic immune activation, including the systemic availability of intestine-associated microbial products. Both activating NKG2C and inhibitory NKG2A receptors recycle and are expressed on the cell surface as heterodimers with the CD94. IL-12 was recently shown to promote the upregulation of the CD94-NKG2A complex on NK cells, thereby modulating the NKG2C-dependent responsiveness. 14 Further cytokines, including IL-15, promote the maintenance of CD56 and activating NCR, NKp30, and NKp46. 15 Both IL-12 and IL-15 are diminished during HIV-1 infection and this loss may favor a phenotypic shift toward a CD56−CD16+NKG2C+NCRdull/- phenotype, likely contributing to NK cell dysfunction. 16,17 The ratio of NKG2A:NKG2C+ NK cells is inverted in CMV-seropositive, HIV-1-infected individuals with severe disease and is not normalized until after 24 months of antiretroviral therapy. 13 The duration of the altered NK cell phenotype could account for the maintenance of elevated NKG2C expression in aviremic HIV-1+ individuals receiving HAART shown here, despite this group having higher CD4+ T cell counts than HIV-1+ viremic groups.
Previous studies have shown that HCMV-seropositive HIV-1-infected individuals have significantly higher proportions of NKG2C+ NK cells. 9,10 We observed a weak correlation between anti-CMV IgG plasma antibody titer in individuals with high HIV-1 viral loads and either the proportion of CD56−CD16+ cells or NKG2C+CD56−CD16+ cells, possibly reflecting a direct role of HCMV in driving dysfunctional NK cells. However, there was little difference in the proportions of CD56−CD16+ cells or NKG2C+CD56−CD16+ cells between HCMV-seronegative and HCMV-seropositive HIV-negative control individuals, indicating that the frequency of reactivation, CMV viral titer, and HIV-1 coinfection are likely to play a role in driving the expansion of these subsets. Four HIV-1-seropositive individuals were HCMV seronegative, all of whom were receiving antiretroviral therapy with HIV-1 viral load <50 copies/ml blood. A nonsignificant trend toward a lower frequency of CD56−CD16+NKG2C+ cells was observed in HCMV-seropositive compared to HCMV-seronegative individuals within this group, indicating a potential influence of CMV exposure (p=0.107, data not shown).
Recent studies of umbilical cord stem cell transplantation indicate that cytomegalovirus reactivation promotes an accelerated NK cell maturation associated with the expansion of CD56−CD16+NKG2C+ cells. 18 Mechanistically, such an expansion of NKG2C+ cells could be accounted for by the stabilization of HLA-E on the surface of APC by HCMV-derived peptide sequences, providing an activating signal for this NK cell subset of NK cells. 19 –21 This is borne out by induction of proliferation in NKG2C+ NK cells by HCMV-infected fibroblasts. 22 Further work on the contribution of concomitant infections to chronic NK cell activation and dysfunction will be of importance in the development of strategies to combat immune exhaustion during HIV-1 infection.
Footnotes
Acknowledgments
This work was funded by the St Stephens AIDS trust and by the Joint Research Committee of the trustees of Chelsea and Westminster Hospital. M.R.G. is funded by the Campbell Foundation and the Medical Research Council (UK).
Author Disclosure Statement
No competing financial interests exist.