
Abstract
Among persons infected by HIV-1, the rate of progression to AIDS is multifactorial being affected by host and viral factors, including the HIV-encoded negative factor (Nef). Our aim was to define whether variations in the nef gene as well as its functions may be associated with slower HIV disease course in infected children. The proviral HIV-1 nef gene was cloned, sequenced, and compared in children with contrasting disease course: 10 long-term nonprogressors (LTNP) and six rapid progressor (RP). The CD4 and MHC-I down-modulation ability of nef alleles derived from LTNP and RP children was analyzed. We observed that only one of our 10 LTNP had a protective genetic background, and out of them, 40% had defective nef genes, carrying substitutions at the (AWLEAQ56–61) and the (Rxx22–24) domains, and that those alleles were unable of down-regulate CD4 and MHC-I. The emergence or presence of Nef L58V substitution was associated with viral attenuation, indicated by a reduction in HIV viral loads, a persistent preservation of CD4+ T cell counts, and lack of AIDS-related symptoms. Our results demonstrate that HIV-1 perinatally infected children carrying functionally defective nef HIV-1 strains have prolonged asymptomatic phases without therapy, suggesting a relevant role of CD4 and MHC-I down-modulation Nef domains on in vivo HIV-1 pathogenesis and pediatric immunodeficiency outcome.
Introduction
AIDS
HIV-1 Nef, a 27- to 32-kDa myristoylated protein, has been considered one of the viral factors that play a major role in HIV pathogenesis in vivo. Nef is highly expressed during the early phase of the viral replication cycle and though it is dispensable for HIV-1 replication, it can enhance viral infectivity in vitro by at least 5-fold. 12 This protein has multiple effects on the immune system including cell surface down-modulation of CD4 and major histocompatibility complex class I (MHC-I A and B) on infected cells. 13,14 CD4 down-regulation prevents HIV-1 superinfection and allows virion budding from infected cells. 13,15 HLA-A and HLA-B down-modulation permits infected cells to escape from specific cytotoxic T lymphocytes (CTL). 14 Moreover, Nef is able to bind host cell protein kinases such as Lck, Hck, Akt, Alix-1, and others, modulating many cell metabolic routes. 14
Experimental studies using transgenic mice expressing the nef gene have shown lymphocyte depletion and AIDS-like-associated pathology. 16,17 On the other hand, Rhesus macaques infected with SIVΔnef, with an attenuated replication capacity and pathogenicity, had preserved CD4+ T lymphocyte counts with low viral loads. 18 Collectively, these data suggest that Nef is an important virulence factor preventing immune system recognition of infected cells, enhancing virus replication, infection, and virulence. 18,19
Many studies have shown an association between defective nef gene and slow or nonprogression of HIV infection, 20 –32 mainly in cohorts of adults, though few have been focused on the association of the nef gene with disease course in HIV-1 vertically infected children. 29 –31
The aim of our study was to explore the role of the nef gene in pediatric HIV-1 infection selecting a unique group of patients with polar clinical outcomes. We found that punctual mutations could impact Nef functions and attenuate HIV-1 pathogenicity in vivo reflected by viral loads and preserved CD4 T cell counts in long-term nonprogressors (LTNP) children.
Materials and Methods
Patients
Sixteen infants were included in the study, selected due to their clinical characteristics and laboratory parameters (Table 1). All of them were diagnosed with vertical HIV-1 infection at birth or within the first months of life by multiplex polymerase chain reaction (PCR). Ten of them remained asymptomatic with preserved CD4+ T cell counts, without receiving any antiretroviral (ARV) therapy for at least 8 years, fitting the criteria for LTNPs as defined elsewhere. 29 LTNPs #3467 and #3468 were siblings born to the same mother. We also included six rapid progressor infants (RP) who developed AIDS within 2 years of infection. Informed consent from their parents or legal guardians was obtained and the study was approved by the Institutional Ethic Committee of the Pediatric Hospital “Prof. Dr. Juan P. Garrahan” of Buenos Aires City.
Mean±SD of all available data from each patient. The clinical and laboratory follow-up for all but two (#4273 and #2074) of these patients was every 6 month since they were diagnosed. Plasma viral loads were determined using Branch DNA (ORGANON), COBAS Amplicor (ROCHE), or COBAS Taqman (ROCHE) methods. CD4+ T cell counts were assessed by flow cytometry staining for CD3/CD4 and determining the percentage of CD3/CD4+ cells.
LTNPs who have never received ARV therapy until the present time.
Siblings.
Only one sample was available for this LTNP.
Patients #193 and #38 died at 8 and 12 month, respectively; we had data on CD4+ T cell counts and VL from only one sample of each one.
ARV, antiretroviral; LTNP, long-term nonprogressor; RP, rapid progressor.
HLA-B genotyping
HLA-B alleles were genotyped at the molecular level by using a fluorescent bead-based assay using the Luminex platform (Luminex, Austin, TX) for high resolution. In brief, the LIFEMATCH_System (Gen-Probes, Stamford, CT) for HLA-B typing is based on the simultaneous detection of multicolored beads in suspension. In our study, one tube reaction containing the PCR-amplified specific HLA-B product was hybridized with a set of probes attached to the fluorescent beads and discrimination of positive hybridization was allowed by the use of streptavidin-phycoerythrin binding to PCR products carrying original biotin-labeled primers. HLA-B alleles were assigned by using the LIFEMATCH 2.5.2 software provided by the company.
CCR5 and CCR2 genotyping
All patients were analyzed for CCR5Δ32 and CCR2 64I alleles. CCR5 were studied by PCR looking for the fragment 32 bp smaller (CCR5Δ32), and CCR2 64I were identified by PCR-RFLP using the enzyme BsaBI as previously described. 6
nef gene amplification, cloning, and sequencing
The proviral nef gene was amplified directly from peripheral blood mononuclear cell-isolated DNA by nested PCR using Taq DNA polymerase (Invitrogen Life Biosciences). The outer primers were NefF1 (5′-GCAGTAGCTGAGGGGACA GATAG-3′) and NefR1 (5′-CCAGTACAGGCAAAAAGC AGCTGC-3′), corresponding to nucleotides 8676–8697 and 9530–9507 in the HIV-1 NL4-3 genome, respectively. The inner primers were NefF2 (5′-GCACATACCTAGAAGAAT AAGACAGG-3′) and two alternative reverse primers, NefR2 (5′-GCCACGCCTCCCTGGAAAGTC-3′) that amplified clade B nef genes selectively and another primer designed using the Argentinean BF-CRF prototype sequence ARMA159 (GenBank: AF385936) due to the high frequency of that type of CRF in the Argentinean and South American pediatric population 33 : NefRSeq (5′-CCCGCCCTCTGGAAAGTC-3′). PCR products from two independent amplifications from the same cell sample were mixed and purified from agarose gel using a Perfectprep Gel Cleanup Kit (Eppendorf AG 22331, Hamburg, Germany). When the amplification pattern showed more than one band in 2% agarose gel, the purified and sequenced band was always the band of the expected 680 bp size.
Purified fragments were cloned using the TOPO easy vector kit (Invitrogen, USA) according to the manufacturer's protocol. Recombinant vectors were used for transforming competent DH5α E. coli and 10 white colonies were picked up from those plates containing more than 30 white colonies but less than 300 colonies. The ligation efficiency was always higher than 70%. The amplified cultures were plasmid purified and each clone was sequenced using the T7promoter and M13reverse universal primers with the DYEnamic ET Terminator Cycle Sequencing Kit (Amersham Biosciences, UK Limited) on an ABI PRISM 3130 sequencer (Applied Biosystems, USA) according to the producer's protocols.
Sequence analysis
The forward and reverse nef sequences from each sample were aligned using MEGA 4.0 software by Clustal X and refined manually. The corrected sequences were defied with Basic BLAST blastn (
Alignments with subtype reference sequences from the Los Alamos database (available at URL:
CD4 and MHC-I down-modulation analysis
The Nef ability to down-regulate CD4 and MHC-I was assessed as reported previously by Turk et al.
36
Briefly, each selected nef allele was subcloned into pCG-IRES-GFP vector (obtained from Dr. Benaroch's laboratory) in order to express Nef and GFP bicistronically. The recombinant vectors were used to transfect HeLaP4 cells using Lipofectamine2000 (Invitrogen, USA). The transfected cells were incubated 24 h at 37°C, 5% CO2 in H-DMEM added with 10% FBS, 2 mM
Results
Host genetic factors: HLA-B, CCR5, and CCR2
We first evaluated if host genetic factors previously associated with HIV-1 disease progression accounted for the clinical differences in LTNP and RP children of our cohort. Genetic variations in HLA-B, CCR5, and CCR2 are illustrated in Table 2. Homozygosity for HLA-B alleles, a factor previously associated with rapid disease progression, was not found in any of the 14 patients. Data from RPs #193 and #104 could not be obtained because of lack of DNA samples. The siblings #3467 and #3468 had identical HLA-B alleles. Two of the slow progressors (#4273 and #509) had the HLA-B*5701 allele, linked to control of HIV-1 infection. Remarkably, three out of four RP typified (#38, #12, and #92) but none of the LTNP were positive for HLA-B*35, associated with rapid disease progression, with the association between HLA-B*35 and rapid progression to AIDS statistically significant (data not shown). Furthermore, patient #12, in addition to B*35, carried the B*0801 allele, also associated with rapid progression. No association between HLA-B alleles and relevant mutations within the nef gene was found in patients with slow HIV-1 disease progression. Only one (#509) of the LTNP carried the CCR5Δ32, and none of them had the CCR2 64I allele (Table 2). RPs #3 and #104 were heterozygous for CCR5Δ32 and CCR2 64I, respectively.
WT, homozygous wild type.
Siblings.
Heterozygous CCR5Δ32.
ND, not determined.
Heterozygous CCR2 64I.
HIV-1 Nef variants in LTNP and RP children
To determine the frequency of nef-defective HIV-1 strains in vertically infected children and to evaluate its association with the rate of disease progression, nef genes were amplified, cloned, and sequenced from two groups of pediatric patients, 10 LTNP and six RP. We found that in four LTNP subjects (#513, #3467, #4273, and #3070), all the nef gene clones carried mutations at one functional site (Table 3). Three of these subjects (#513, #3467, and #4273) carried mutations within the AWLEAQ56–61 site, and, interestingly, patients #513 and #3467 had exactly the same mutation (Table 3). Briefly, six out of seven clones from #513 had the substitution L58V, while the remaining clone was truncated. Mutation L58V was also observed in all the clones (n=10) from patient #3467, but three of them also had a premature stop codon (W57Stop). In the remaining patient (#4273) with mutations within the AWLEAQ site, the nonconservative change A60E was observed, changing a neutral residue for a glutamic acid. In patient #3070, the Rxx22–24 was absent in all the clones (n=10) that had the substitution R22Q (Table 3), suggesting that this HIV-1 variant might be unable to down-regulate HLA-A and HLA-B. The mutation R22Q was also observed in half of the clones from two LTNPs (#509 and #513) and in one RP child (#104). As shown in Table 3, other relevant substitutions were exclusively found in the three LTNP that had the AWLEAQ56–61 site mutated.
Clones detailed in the table are all from the same sample of each patient.
Length variations with respect to NL4-3 laboratory strain nef gene.
BF, recombinant form between clade B and clade F that carried a recombination point within the nef gene.
The remaining clone was truncated due to a not-in-frame deletion, harboring a premature stop codon.
Three clones had a premature stop codon (W58Stop); inspite of this, the codon corresponding to residue 59 was always translated as valine.
Three of the six clones mentioned were also truncated (W58Stop).
Siblings.
Also in-frame insertions or deletions located within the amino-terminal loop were found. These length polymorphisms were observed in clones from seven patients with slow progression and only in the RP #3 (Table 3). Sequences bearing insertion/deletion were found as unique length variants in all the clones from LTNPs #41, #3182, and #4273 and for the RP #3, or as an additional length variant in LTNPs #3467, #513, #2074, and #509. Not-in-frame deletions/insertions were observed in only nine out of 146 clones (4/89 from LTNP and 5/57 from RP).
In addition, to determine nef gene HIV-1 subtype and phylogenetic relationships between them, clone sequences from all patients were aligned and analyzed together with group M subtype references in a neighbor-joining tree (Fig. 1). The analysis allowed us to discard intersample cross-contamination and to establish that mutations found in LTNP were not associated with a particular HIV-1 subtype and did not have a common origin (Fig. 1, Table 3).
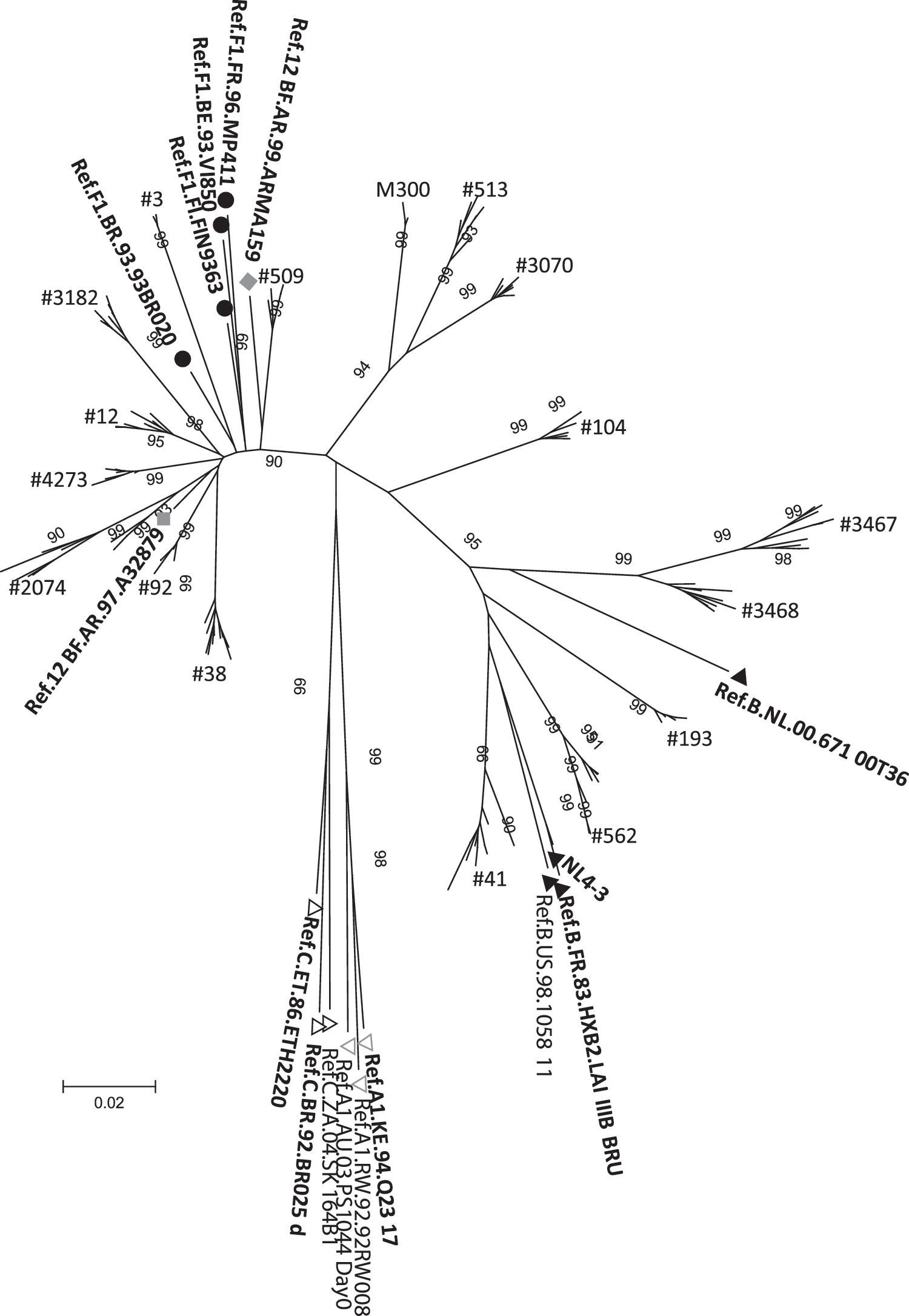
Evolutionary relationships of sequences obtained from the 10 long-term nonprogressors (LTSP), six rapid progressors (RP), and mother of patient #513 (#M300). The evolutionary history was inferred using the neighbor-joining method. The optimal tree is drawn to scale, with branch lengths in the same units as those of the evolutionary distances used to infer the phylogenetic tree. The evolutionary distances were computed using the Kimura two-parameter method and are in the units of the number of base substitutions per site. The rate variation among sites was modeled with a gamma distribution (shape parameter=1). All positions containing gaps and missing data were eliminated from the dataset (complete deletion option). A total of 519 positions were in the final dataset. Phylogenetic analyses were conducted in MEGA4.0. Each cluster was named as each patient and references names are shown in the tree. Black circles indicate subtype F1 references, black triangles indicate subtype B references, gray diamonds indicate CRF_12 BF references, gray line triangles indicate subtype A1 references, and black line triangles indicate subtype C references.
Modulation of immune receptors CD4 and MHC-I from the cell surface
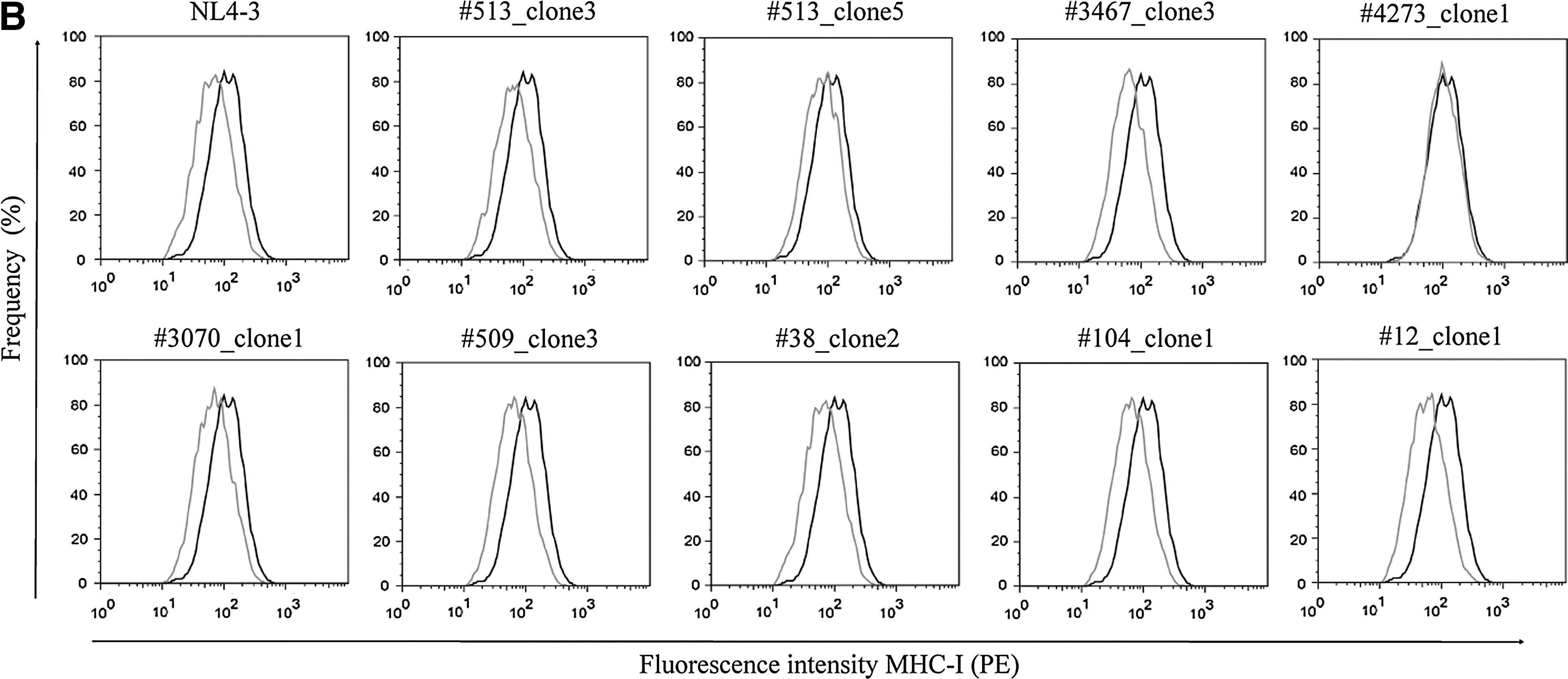
To study how Nef polymorphisms impact Nef activity, the capacity of eleven different Nef alleles to modulate cell surface immune receptors was evaluated (Table 4). Assayed alleles were derived from LTNPs and from RPs, selected on the basis of carrying or not carrying relevant substitutions at functional motifs. We observed that alleles having the substitutions described above (Table 3) failed to perform the functions associated with such sites (Fig. 2 and Table 4). The combination of mutations as L58V+E59K (#3467_clone3) and A60E+ins33bp (#4273_clone1) resulted in loss of CD4 down-modulation ability (Fig. 2A). The R22Q mutation itself accounted for lack of MHC-I regulation activity (#513_clone5 and #3070_clone2). As well, the allele #4273_clone1 was also unable to down-modulate MHC-I (Fig. 2B). Surprisingly, the allele #104_clone1 derived from an RP, carrying the same mutation, had a lower modulation capacity though the difference with NL4-3 was not significant. Mutation A61T, present in almost half of the clones of RP #38, did not alter Nef functionality (#38_clone2). Alleles having no mutations (#509_clone3 and #3182_clone1 from LTNPs; #12_clone1 and #92_clone2) were as active as the NL4-3 clone performing either CD4 or MHC-I down-regulation (Fig. 2 and Table 4).
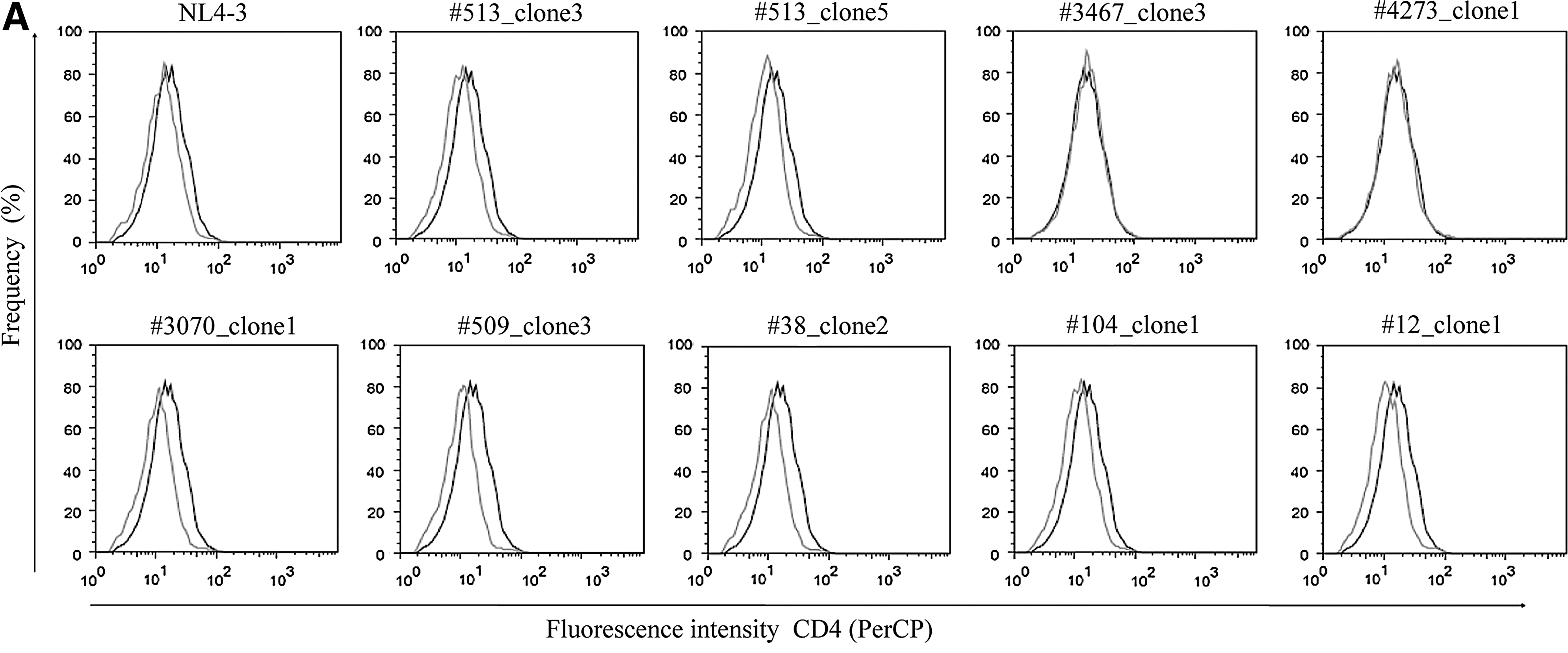
MHC-I and CD4 down-modulation activity of Nef alleles derived from LTNP and RP selected children. Nef alleles are indicated above each graph. Living cells were first gated on an SSC vs. FSC dot plot. Transfected cells (GFP+) were then visualized on a GFP vs. FSC plot. Finally, CD4
Mutations with respect to wild-type NL4-3. Dashes indicate that there were no mutations at the functional sites analyzed.
The difference, though appreciable, is not statistically significant (p=0.07).
, **Statistically significant differences from NefNL4-3 are denoted with one (p<0.05) or two (p<0.001) asterisks.
Discussion
We found that HIV-1 variants bearing defects at conserved Nef domains 14 lacking the ability to down-regulate surface CD4 and/or MHC-I molecules were present in more than one-third of the HIV-1 perinatally infected children classified as LTNPs. Thus, single mutations at functional sites of Nef can attenuate viral pathogenicity in vivo by loss of immunoreceptor surface modulation activity, suggesting that Nef functions may have an influence on the rate of HIV-1 disease progression.
Different reports have demonstrated that gross deletions within HIV-1 nef/LTR region attenuate HIV in vitro. Those kind of defective variants have been found in very few patients, though they were always isolated from LTNPs and never from typical or rapid progressors. Genetic defects within the HIV nef gene have been evaluated by others in around 219 adult subjects with characteristics of LTNP or elite controllers, and only 22 of them carried gross deleted nef strains. Since the nine individuals of the Sidney Blood Bank Cohort represent only one transmitted variant, the proportion of HIV-1-infected adults bearing defective nef genes decreases to 6% (13 of 211), being slightly larger if punctual mutations at conserved Nef sites were included. 2,20,22,24,26,37 –40
In general, children infected by vertical transmission have a shorter asymptomatic phase or survival time than adults. Approximately 20% progress rapidly to AIDS within the first 2 years in the developed world. 41 HIV-1-infected children with natural long-term viremia control and immune function preservation have not been reported. 42 –45 In Argentina, we had the unique opportunity to recruit 10 LTNP HIV-1-infected infants persistently asymptomatic, preserving the CD4+ T cell counts and ARV free for more than 8 years, based on retrospective HIV-1 vertical infection diagnosis since 1986. 46
In the literature, genetic defects within the nef gene were extremely rare in the context of pediatric infection. 29 –31,47,48 Only one LTNP child with a gross nef-deleted strain was documented. 31 Though the frequency of defective nef alleles was significantly higher for slow or nonprogressor children than for RPs, 29 –32,47,48 mutations at conserved functional sites were as rare in children as in adults.
More than one-third of our LTNPs carried exclusively defective nef HIV-1 variants, with mutations at conserved Nef motifs associated with CD4 and MHC-I down-modulation domains. It was demonstrated that Nef-dependent CD4 and MHC-I down-regulation is strongly associated with immunodeficiency outcome and nef alleles derived from LTNP are significantly less efficient for those functions. 22,30,49,50 Rhesus macaques infected by modified SIV strains that are Nef deficient solely in down-modulating surface CD4 do not progress to AIDS-like disease. 51,52 However, reports of the same kind of Nef defects for HIV-1 in vivo are few. 22,48 Carl et al. 22 have described an LTNP case infected by an HIV-1 virus carrying the substitution W57R within Nef, located at the CD4 down-regulation domain (WL57–58), that was indeed unable to down-regulate cell surface CD4. This patient remained asymptomatic with normal CD4+ T cell counts and remained ARV free for more than 15 years.
As well, Zuo et al. 48 recently reported that three out of 15 long-term-survival HIV-1 vertically infected individuals with functional-defective Nef were unable to perform either CD4 or MHC-I down-modulation. Together, these previous data further support the association between Nef-dependent CD4 or MHC-I down-modulation and HIV-1 pathogenicity. In addition, mutations described here (L58V and A60E) are located at the motif implied in Nef-induced bystander CD4+ T cell apoptosis. 53 The disruption of this domain could alter and avoid the Nef-associated “bystander effect” in these patients, preserving Th lymphocyte counts 54 and leading to a nonprogressive HIV-1 infection.
We observed that the L58V mutation could account for the reduction of in vivo HIV-1 replication and pathogenicity. It was observed in all clones from two LTNP unrelated children (#513 and #3467). The longitudinal analysis performed in one of them (#513) showed that the emergence of the L58V was linked to a decline in viral load from high to undetectable levels, remaining undetectable for more than 6 years up to the present time (data not shown). For the other case, comparison of nef sequences obtained from HIV-1-infected siblings #3467 and #3468 revealed an association between Nef and pathogenicity. Though both siblings had identical HLA-B, CCR5, and CCR2 alleles, and were infected with phylogenetically related viruses, only one of them has maintained normal CD4+ T cell counts until the present time and has been ARV naive for more than 15 years. This child was infected by an HIV-1 mutated at the Nef CD4 down-regulation domain. However, the sibling with wild type nef HIV-1 required ARV therapy at 9 years of age due to an abrupt decline in CD4+ T cell counts.
These findings suggest that even with the same virus backbone and host genetic environment, single changes within the nef gene may have an influence on the outcome of pediatric HIV-1-associated immunodeficiency.
Ethnicity, age, HIV subtype, coinfections, and the different study designs in HIV-1-infected children are some of the factors that might account for discrepancies observed between our report and others. All of our cases were white-Hispanic Argentineans and 75% were infected with HIV-1 BF recombinant forms, considering that the nef genes typified as clade F belong to a CRF_BF, an intersubtype almost exclusive to South America. 34 Moreover, the criteria we used to define long-term nonprogressor children, described in Materials and Methods, were different from that used by others, 31 since 4-year-old children were considered slow progressors by these authors. In regard to the study design, we analyzed clone cellular provirus sequences while in some other reports the authors have evaluated free cell virus or provirus by direct sequencing. 31,47 We observed not-in-frame deletions by direct sequencing for samples from LTNPs. None of the clones showed those punctual deletions (data not shown). Discrepancies observed between these sequencing strategies may be due to polymerase errors in polypurine tracts of variable sequences as found in nef genes from LTNPs as was reported elsewere, 48 suggesting that direct sequencing of the HIV-1 nef gene may lead to wrong interpretations.
Since RPs and LTNPs could not be matched by age, we could not establish whether nef defects found in LTNPs were a causal factor for slow progression or a consequence of being infected for more than 10 years.
We also observed a significant association between the HLA-B*35 alleles and rapid disease progression that was previously demonstrated by other authors, 10 although the cohort analyzed is small. The LTNP status of the children included was not a consequence of carrying one of the known host protective alleles. This analysis was not an association study but an additional cohort characterization.
The host genetic background is a factor that was omitted in most reports of Nef/AIDS progression association. It is in fact a confounding factor, since in patients having a protective genetic background, the status of LTNPs will probably reflect it and will not be a consequence of HIV-1 attenuation; thus, the frequency of viral genetic defects could be underestimated. In our cohort, only one child (#509) had a genetic background that was clearly associated with slow or non-AIDS progression (HLA-B*5701+CCR5Δ32).
Our study indicates that Nef plays a relevant role in pediatric HIV pathogenesis and supports the importance of the conserved Nef CD4 and MHC-I down-regulation functions for HIV-1 virulence. Mutations that disrupt any of those Nef domains could be used as predictors for slow AIDS progression. Point mutations are enough for attenuating virus pathogenicity in vivo, since single substitutions lead to a lack of Nef-dependent CD4 or MHC-I down-modulation. HIV-1 variants carrying defective nef genes are not frequent, but individuals infected by those strains have prolonged asymptomatic periods and an attenuated course of HIV-1 infection.
Sequence Data
Sequence accession numbers: GenBank JF283601 to JF283778. Release date was August 3, 2011.
Footnotes
Acknowledgments
The authors are grateful to Dr. Sunil K. Ahuja from the Department of Medicine of the University of Texas Health Science Center, San Antonio, Texas and Dr. Andrea Raymond from the Morehouse School of Medicine of Atlanta for critical reading of the manuscript. We also thank Ms. María del Cármen Galvez, Mrs. Natalia Beltramone, and biochemist Silvia Marino for their excellent technical assistance and collaboration on laboratory handwork.
Author Disclosure Statement
No competing financial interests exist.