
Abstract
Insight concerning the switch in HIV-1 coreceptor use will lead to a better understanding of HIV-1 pathogenesis and host-virus dynamics. Predicting CXCR4 utilization by analyzing HIV-1 envelope consensus sequences is highly specific, but minority variants in the viral population are often missed resulting in low sensitivity. Commercial phenotypic assays are costly, and the development of sensitive in-house phenotypic assays to detect CXCR4-using HIV may not be feasible for some laboratories. A sensitive, inexpensive genotyping assay was developed to detect viral sequences associated with CXCR4-utilizing virus (X4). Codon-specific primer pairs were used to detect X4-associated codons at five positions in the HIV-1 envelope V3 loop (11, 13, 24, 25, and 32). Sixty plasma samples from HIV-1-infected individuals were analyzed by consensus sequencing and codon-specific PCR (CS-PCR). Forty-six of these were also phenotyped by Trofile or Enhanced Sensitivity Trofile (ESTA). CS-PCR detected X4 variants 17% more often than 11/24/25 consensus sequencing alone (n=60), 30% more often than Trofile (n=27), and in a limited data set, 16% more often than ESTA (n=19). CS-PCR combined with consensus sequencing had approximately 80% concordance with ESTA.
Introduction
HIV-1
Detecting and monitoring coreceptor use in a laboratory setting are not trivial processes. X4 subpopulations are often at low levels—particularly early during disease progression—and even though there are multiple, well-established genotypic tools, 2 –5 population-based sequencing can generally detect only variants that are greater than 20% of the viral population. 6 In addition, consensus genotypes can be indeterminate when subpopulations are variable at the codons of interest, or when insertions or deletions cause overlapping reading frames; both are common in the HIV-1 envelope. Next-generation sequencing may prove to be the best method of detection, as it is not only sensitive, but can also give a fairly accurate estimation of the percent X4 in a population. 7,8 However, next-generation sequencing is still fairly expensive and handling the data can be onerous. Phenotyping is another option for X4 detection, but many laboratories do not have the facilities or expertise to develop a sensitive culture-based assay, and commercial assays, such as Trofile, are costly. In addition, multiple amino acids in the HIV-1 envelope protein affect the efficiency of coreceptor use with progressively more changes occurring as the R5 viral population transitions to variants that can use CXCR4. 9 How well a variant can use CXCR4 may influence how well phenotypic assays perform.
CXCR4 use by HIV-1 is associated with viral genotypes that have arginine or lysine at position 11 or 25 in the variable loop 3 (V3) of the HIV-1 envelope protein. 10 The identification of basic amino acids at positions 11 and 25 has >90% specificity for identifying HIV-1 subtype B X4 variants. 2,3 Multiple three-base combinations encode the many amino acids that can be found at positions 11 and 25, including the eight that can encode lysine or arginine. Therefore, no single base can be used to differentiate X4-associated versus R5-associated codons.
This article describes a relatively inexpensive polymerase chain reaction (PCR)-based method to detect X4 by specifically amplifying X4-associated codons found in the V3 loop of HIV-1 subtype B envelope. Codon-specific PCR (CS-PCR) targets five codon positions that most commonly encode one or more basic amino acids in X4 variants (11, 13, 24, 25, and 32). A semiquantitative first round PCR (qPCR) was performed to ensure sufficient input of viral templates from each specimen and the resulting amplicon was used as the template for CS-PCR. The genotypic detection of X4 by CS-PCR combined with consensus sequencing was compared to consensus sequencing alone and to a commercial phenotypic assay (Trofile or Extra Sensitive Trofile Assay).
Materials and Methods
Specimens and human subjects approval
Clinical specimens, as approved by the University of Washington and the Children's Hospital of Philadelphia Institutional Review Boards, were utilized to optimize the assay. Additional test specimens with 454-pyrosequence data, graciously provided by Dr. Richard Harrigan with permission from Pfizer, were used to define cutoffs.
Isolation of RNA and generation of cDNA
Virus was pelleted from 1.0 to 1.5 ml of plasma by centrifugation at 25.2K×g for 2 h at 4°C. The supernate was carefully removed leaving 50–100 μl of plasma undisturbed above the pellet. The volume over the pellet was adjusted to 140 μl with phosphate-buffered saline (PBS) and RNA was isolated using the Qiagen QIAamp Viral RNA Mini Kit (Valencia, CA) as per the manufacturer's protocol.
cDNA was generated using 5 μl of the extracted RNA in a 20-μl reaction containing 10 pmol of ED12 primer (Table 1), 500 μM dNTP, 1 μl 0.1 M dithiothreitol (DTT), 0.5 μl RNasin (Promega, Madison, WI), and 1 μl of Superscript III (Invitrogen). Primer, RNA, and dNTP were heated at 65°C for 5 min, cooled to 45°C for 5 min, at which time the enzyme, buffer, DTT mix were added, and incubation was continued at 50°C for 1 h.
F, forward primer.
R, reverse primer.
pmol/RX, pmol of each primer per reaction.
TMT or MGT refer to the degeneracy added to the reverse primer mix to accommodate variability at codon 13.
LNA, locked nucleic acid, are indicated in parentheses.
Quantification of input virus by real-time PCR
The cDNA was amplified using semiquantitative real-time PCR with two primer sets (Table 1) in a BioRad iCycler (Hercules, CA), each in duplicate. Two different primer sets were used to increase the likelihood of efficient amplification of envelope sequences. Each 50 μl reaction was prepared with 0.5× SensiMix (Sybr Green-fluorescein formulation, Bioline, London, UK) and 15 pmol each primer. To compensate for using only half concentration real-time mix, the reaction mixture was adjusted to 1× buffer with Bioline Biolase buffer (London, UK) and an additional 75 nmol magnesium and 5 nmol dNTPs were added. Two to 5 μl of cDNA was added to the mix and the reactions were carried out as follows: 10 min at 95°C; followed by 50 cycles of 10 s at 92°C, 20 s at 55°C, and 20 s at 72°C concomitant with Sybr Green detection. Following the last cycle a melting curve was generated from 60° to 100°. The p2-7 plasmid (Table 2) was used to create a standard curve (104, 103, 102, and 10 copies). The quantified input template from each of the four reactions was totaled (2 replicates×2 primer sets). If <100 copies of input cDNA were amplified, the cDNA/qPCR was repeated to amplify a minimum of 100 copies of cDNA, or until the sample was exhausted.
Designates whether the codon is X4- or R5-associated and is accordingly used as an X4 or R5 control.
CS-PCR and generation of the consensus sequence
The qPCR from each sample was combined in a single tube and 100 μl of the mixed amplicon was purified (QIAquik PCR Purification Kit, Qiagen). DNA was eluted with 50 μl elution buffer, quantified using a spectrophotometer (Nanodrop, Thermo Scientific, Wilmington, DE), and normalized to 30 ng/μl by the addition of Tris-EDTA buffer (TE). Five microliters of the normalized template was visualized in an agarose gel to verify that the PCR products were of the desired size and of similar intensity across specimens. A consensus sequence was generated using 0.5 μl of DNA in a 5 μl Big-Dye (ABI, Carlsbad, CA) sequencing reaction with primer envC2.F2 and/or envC3.R1 (Table 1).
Each normalized template was diluted 1:200 in TE buffer resulting in ∼3×108 templates/μl (performed in strip-well tubes for efficient transfer with a multichannel pipette) and was then subjected to CS-PCR, where each codon was amplified individually. CS-PCRs were performed in 25 μl containing 1 μl of diluted template in 0.5× SensiMix and 7.5 pmol of each primer (Table 1). The reaction mix was adjusted to 1× buffer by adding Bioline Biolase buffer (without magnesium). PCR conditions were 95°C for 10 min, followed by 44 cycles of 10 s at 92°C, 1 s at either 45°C (codons 11, 13, 24AGA, and 25AGA) or 40°C (codons 24AAA, 25AAA, and 32) and 5 s at 72°C (concomitant with Sybr detection). This was followed with a melting curve from 60° to 90°C.
CS-PCR primers, controls, and set-up
Codon-specific primer pairs were designed to detect one or more X4-associated codons at positions 11, 13, 24, 25, and 32 of the V3 loop (c11, c13, c24, c25, and c32) (Table 1). Primer pairs bisect the target codons in one or two configurations (NN+N and/or N+NN, Fig. 1a and b) except for c11 CGT, which overlaps at the first base of the codon (Fig. 1c). To increase amplification specificity, the primers have “engineered” mismatches introduced at the second or third base from each 3′ end. Each CS-PCR primer pair has matching X4-codon and consensus R5-codon control plasmids (Table 2). A 1:100 mixture of X4:R5 controls was used to monitor sensitivity. Plasmid controls were linearized with EcoRI and diluted to ∼3×108 copies/μl.

Primer designs for codon 11 AGG (arginine), codon 11 CGN (arginine), and the codon 11 sample-amplification-control. Codon 11 is underlined by a bold black bar. Primer sequences are shown in bold and the engineered mismatches 2 or 3 bases from the 3′ end are marked with an asterisk.
To monitor sample input concentration and gauge the effect of primer mismatches, the normalized sample templates were also amplified by primer sets that precisely flank c11 and c25 (Fig. 1d). These reactions are referred to as sample-amplification-controls (SAC).
Typically, 10 viral samples were tested with three codon-specific primer pairs and the sample-amplification-control primers, all in duplicate, in a 96-well plate (see Fig. 2). Each set of codon-specific reactions was accompanied by the respective controls: X4, R5, 1:100 mix of X4:R5, and a no DNA. Each X4 and R5 control was also amplified by the sample-amplification-control primers to monitor three parameters: whether too much sample template was submitted to CS-PCR (which was indicated by an earlier real-time cycle threshold than the R5 control), the stability of the controls, and the consistency of assay performance across experiments. These are referred to as X4 and R5-amplification-controls (XAC and RAC).
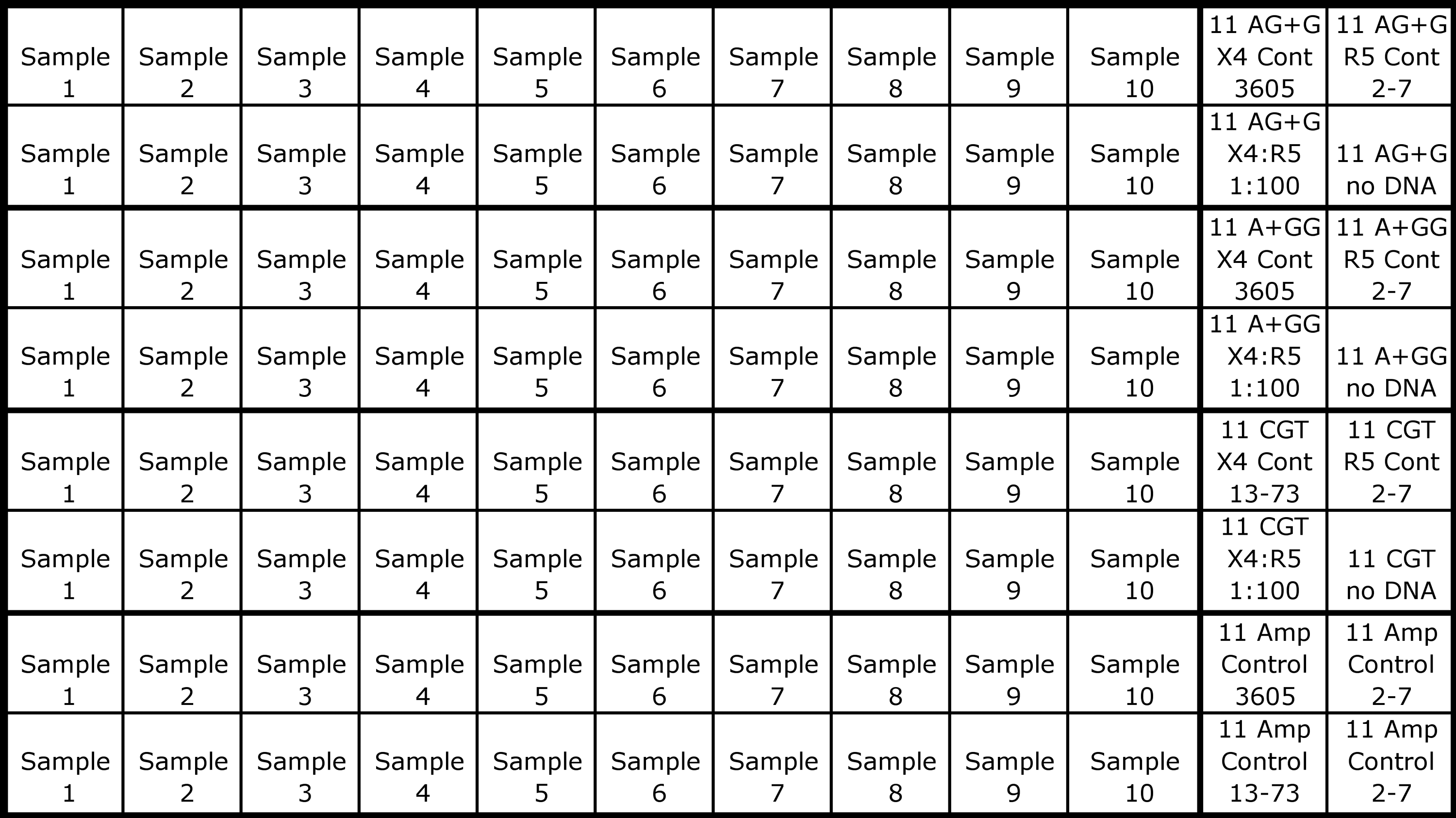
Example plate format for codon-specific (CS)-PCR. This configuration tests 10 samples with three codon-specific primer pairs and the sample-amplification-control primers, all in duplicate, in a 96-well plate. Controls for each primer set are in the two right columns. These include the X4 and R5 controls (Cont), the 1:100 X4:R5 sensitivity control, and a no DNA control. In this example, the top two rows screen for c11 AG+G, the second two for c11 A+GG, the third two for c11 CGN, and the bottom two rows contain the amplification controls (Amp Control) for both the samples and the control plasmids. The specific X4 and R5 codon controls for the c11 AGG and c11 CGN codons (see Table 2) are noted in the two right columns.
Interpretation of consensus sequences and codon-specific PCR
Consensus sequence data were edited using Sequencher 4.2.2 (Gene Codes Corporation, Ann Arbor, MI). In chromatograms with low background, the secondary peaks ≥20% of the upper peak were called genotypic mixtures. When background was high, usually due to subpopulations with insertions/deletions, only the highest peaks were recorded. All possible amino acid sequences were subjected to webPSSM (
CS-PCR data were analyzed using the iCycler software. The analysis mode was set to “PCR Base Line Subtracted Curve Fit,” baseline cycles were autocalculated, and the threshold position for each real-time PCR run was set to 50 RFU. The average cycle threshold (C t) value for each sample was subtracted from the C t value for its matching R5-codon control (ΔC t). Positive ΔC t values were recorded. Melting curves were monitored to ensure that a positive ΔC t was not due to mispriming.
The CS-PCR cutoffs were optimized by testing 78 first round env PCR amplicons (termed “test specimens”) that had population sequences and 454-pyrosequences previously generated at the British Columbia Centre for Excellence in HIV/AIDS (Vancouver, Canada). These test specimens were from patients who entered the MOTIVATE/1029 trials for maraviroc after the 1029 study filled up. Processing of the clinical specimens and generation of the HIV envelope PCR amplicons were previously described.
8
Prior to CS-PCR, the first-round PCR products were quantified using a double-stranded DNA quantification reagent (Quant-It PicoGreen; Invitrogen) and normalized to ∼3×108 amplicons per μl. One microliter of the normalized template was amplified by real-time CS-PCR, in duplicate, as described above. CS-PCR results were compared to the most common X4-associated and most common R5-associated sequence obtained from each specimen by 454-pyrosequencing (using a geno2pheno cutoff of 3.5). The CS-PCR results for the test specimens, as well as corresponding % X4 by 454-pyrosequencing and geno2pheno classifications by Sanger sequencing (using a cutoff of 5.75), are presented in Supplementary Table S1 (Supplementary Data are available online at

Decision outline used to analyze the consensus sequence and CS-PCR for the X4-associated genotype.
ΔC ts for the matched AR+R and A+RR reactions for codons 11 and 25 are combined to make the cutoff.
Seventy-seven samples that had previously been genotyped based on 454-pyrosequencing data were assayed by codon-specific (CS)-PCR. Codon/sequence-specific effects on CS-PCR efficiency were noted and ΔC t cutoff values were assigned accordingly.
Statistical comparisons
The detection of X4 viral variants as determined by CS-PCR was compared to genotyping by consensus sequencing alone, and CS-PCR combined with consensus sequencing was compared to X4 classification by the phenotypic assays Trofile and Enhanced Sensitivity Trofile Assay (ESTA) (performed at Monogram Biosciences) using McNemar's two-tailed test. Additionally, the degree of association (kappa) was determined for the comparison with ESTA.
Results
Development of quantitative PCR
To ensure that a sufficient percentage of the viral population was assayed, a qPCR was developed to estimate the amplifiable viral RNA templates isolated from each plasma sample. Subsequently, amplicons derived from at least 100 cDNA templates were submitted to CS-PCR amplification, which ensured sufficient copies of HIV-1 to allow detection of X4 variants at a prevalence of 1–3%. The primers used to quantify the HIV-1 env V3 templates were designed to anneal to the relatively conserved regions flanking V3 (C2, C3, and C4). To increase the likelihood of efficient primer binding, two different qPCR primer mixtures were used in separate reactions. Each reaction contained the same two forward primers along with a unique reverse primer (Table 1). There were no mismatched bases between the primers and the control plasmid used to generate the standard curve (p2-7). Most samples amplified more efficiently with one or the other primer set indicating that mismatches between virus and the primers influenced copy number determination. However, since the standard control matched the sequence of the primers exactly, the qPCR should not have overestimated the amplifiable virus from the samples. Estimates of the number of viral RNA templates amplified from each of the 60 optimization specimens are shown in Table 4.
The genotype is assigned by the occurrence of arginine or lysine at positions 11 or 25, or at 24 plus 13 or 32 in the consensus sequence.
The genotype is assigned by the detection of arginine or lysine at positions 11 or 25, or at 24 plus 13 or 32 using CS-PCR.
The genotype is assigned by G2P3 using the consensus sequence. The false-positive rate was set to the “Optimized cutoffs” based on analysis of clinical data from MOTIVATE (2% and 5.75% FPR). In some cases a secondary sequence with an indel could be discerned with some confidence and was also evaluated by G2P.
The genotype is assigned by PSSM2 using the consensus sequence. In some cases a secondary sequence with an indel could be discerned with some confidence and was also evaluated by PSSM.
Phenotype determined at Monogram Biosciences. The first 32 samples were assayed by the original Trofile and the remainder by the Enhanced Sensitivity Trofile Assay (ESTA).
Specimens from 60 HIV-1-infected individuals (rows 1–60) were examined for X4 virus. The plasma viral loads (VL) obtained clinically are shown in the second column, and the minimum copies of HIV-1 RNA assayed by codon-specific (CS)-PCR and consensus sequencing are in the third column. Values shown in columns 4–17 are the ΔC ts between a positive CS-PCR and its corresponding R5 control (only those ≥2 cycles are shown unless they add support to an X4 classification). Those ΔC ts that are highlighted in darker gray indicate that the codon was also visible in the sequence chromatogram.
seq, an X4-associated codon could be visualized in the sequencing chromatogram, but was not detected by CS-PCR; FrSh, the consensus sequence had a combination of insertions and deletions that made it impossible to analyze by PSSM; Nonrep, nonreportable designation was assigned by Monogram Biosciences; D/M, dual-mixed. Indicates that both coreceptors were utilized by the viruses in the sample. D/M is equivalent to a genotype of X4.
Development of CS-PCR for detection of X4-associated codons c11 (AGG/AGA/CGN) and c25 (AAA/AGA)
The PCR was designed to target whole codons with “codon-specific” primer pairs. The forward and reverse primers bisect the codon such that all three bases of the targeted codon must be present for efficient amplification to occur (Fig. 4). The codon was split A+RR or AR+R, utilizing two sets of primers. One configuration was typically more efficient and/or specific. The c11 CGN arginine primers were designed differently to accommodate all four CGN codons in a single reaction (Fig. 1c). Not all possible codons for arginine and lysine were included in the assay because three codons represent 99% of the basic residues at c11 and two codons represent 95% of X4 variants at c25 (tabulated from an alignment of 6805 HIV-1 subtype B sequences obtained from the Los Alamos National Laboratory HIV-1 Sequence Database).

Codon-specific primers by design do not amplify R5-associated codons. The primers designed to amplify X4 codons are shown in bold with the engineered mismatches marked with an asterisk. Representative double-stranded R5-associated sequences are shown in regular type with the target codon underlined. Mismatches between the sequence and the primers are indicated by vertical lines drawn through the mismatched bases. The primers are designed so that extension is inhibited in at least one direction, hindering amplification. An “X” indicates where extension is impeded and an arrow shows where extension can occur.
c24 (AAA/AGA), c32 (AAA/CGA), and c13 (CGT)
Based on multiple observations that basic amino acids at c24, c13, or c32 are associated with X4 variants, 1,11 –15 these codons were included in the assay, but any one alone was not sufficient to classify the sample as X4. Codons 24 (AAA and AGA), 13 (CGA), and 32 (AAA and CGA) are detected with single reactions in the N+NN configuration.
Discrimination between X4 and R5-associated codons
During development of CS-PCR, multiple primer pairs and reaction conditions were tested in an attempt to balance primer binding at low stringency—which would better accommodate the high degree of HIV-1 envelope diversity—with the tendency of Taq polymerase to indiscriminately extend from the 3′ end of a primer that was not complimentary to the template. Mismatches were engineered at the −2 or −3 position of each primer to increase the specificity of the reaction. This created two mismatches with nontargeted codons at or within one base of the primer's 3′ end (Fig. 4), which greatly decreased nonspecific amplification. The base chosen to introduce a mismatch in the primer was rarely found at that location in sequences obtained from the Los Alamos National Laboratory HIV-1 Sequence Database.
In addition to the engineered mismatches, a low magnesium concentration was required to inhibit nonspecific extension by Taq. The 1× SensiMix real-time mix has 3 mM Mg2+. Half concentration of the real-time mix (1.5 mM Mg2+) increased the specificity thereby reducing false-positive reactions. In addition, the lower Mg2+ concentration decreased the incidence of primer-dimer at low annealing temperatures. Although use of a diluted mix detracts from the simplicity offered by a commercially prepared mix, it halves the amount of an expensive reagent.
Reaction conditions that were sufficiently stringent to completely stop amplification of the R5 controls generally severely reduced the amplification efficiency of the X4 controls. Therefore, optimal reaction conditions were empirically established that resulted in at least a 10 cycle ΔC t between the X4 and R5-codon controls. ΔC ts were calculated for each sample CS-PCR that crossed the threshold before the R5-codon control. ΔC t cutoff values for each codon were established on a “training set” of test specimens that had accompanying 454-pyrosequencing data (Table 3 and Supplementary Tables 1 and 2). 11 ΔC t values for CS-PCR that were greater than the cutoff were considered positive for that codon and X4 determinations were assigned accordingly.
Initially, differentiation of c25 GAA (R5) from c25 AAA (X4) was problematic, with only a few cycles difference between the X4 and R5 control C
t values. To increase the specificity of the c25 AA+A primer set, a “locked” nucleic acid (a modified nucleotide with the conformation fixed to increase hybridization specificity
16,17
) was incorporated at the −2 position of the forward primer, which corresponds to the first G of the R5-associated
Optimization of CS-PCR using clinical specimens that were also analyzed by consensus sequencing and Trofile
Overview of CS-PCR and sequencing results
Sixty plasma specimens from HIV-1-infected individuals were analyzed by CS-PCR and consensus sequencing during development of the assay (Table 4). These samples are separate from the test specimens used to establish the ΔC t cutoffs. The V3 consensus sequence from 34 of the 60 samples (55%) had minority multivariant or indel subpopulations that could not be resolved by the sequence analysis software. Inspection of these chromatograms revealed indel variants in 13 of the 34, including four (#9, 17, 24, and 28) with peaks that had heights greater than 20% of the primary variant.
Discordant results between the two primer sets for c11 or c25
There were discordant results between the two primer sets targeting the same codon (A+RR and AR+R) in 31 (66%) of the 47 reactions that detected X4-associated codons at c11 or c25. In a minority of cases (n=3) mismatches between the viral target and one of the primers was evident in the sequencing chromatogram. For example, c11 in sample #52 was 100% AGG in the consensus sequence. While the c11 A+GG reaction had a high ΔC t, the c11 AG+G reaction was negative due to GTA at c12 rather than the consensus ATA. This substitution created a double mismatch at the second and third bases of the reverse primer in the AG+G reaction, but only a single mismatch at the third base in the A+GG reaction. As another example, testing of sample #49 revealed that c25 GCA could misprime with the c25 AA+A primer set, leading to a false-positive discordant result.
When the paired CS-PCR were discordant, the sample was not typed as X4 unless the combined ΔC t for the paired set was at least four cycles. This was done to decrease the number of false-positive reactions that might be due to alternate or less stringent primer binding with the engineered mismatch.
Comparison of CS-PCR, position-specific scored matrices (PSSM), Trofile, and ESTA
CS-PCR alone detected X4-associated sequences in 25 of the 60 samples (Table 4). Three additional samples were classified as X4 based on a genotype revealed by consensus sequencing. These were not detected by CS-PCR due to multiple mismatches or deletions within the primer binding sites. In all, 28 samples were classified as containing X4-associated sequences.
Genotypic classifications of viral samples using the 11/24/25 rule, geno2pheno, and PSSM are shown in Table 4. The presence of a basic amino acid at codons 11, 24, and/or 25 (i.e., the 11/24/25 rule) 10 in the consensus sequences resulted in classification of 15 samples as X4. Twelve of the 15 codons were detected by CS-PCR. Analysis of the consensus sequences by PSSM, which does not classify sequences based strictly on the sequence at 11/24/25, classified 12 samples as X4. All of these had X4-associated codons by CS-PCR except for sample #24.
Twenty-seven of the 60 samples had phenotypic testing by Trofile (which is reported to have 85% sensitivity when X4 comprises 5% of the population using mixtures of clonal specimens 18 ). Of the 27, only three were found to harbor X4 variants (Tables 4 and 5). Combined CS-PCR and 11/24/25 consensus sequencing typed these three and 10 of the remaining 24 as X4. Specimens from 19 subjects had phenotypic testing by the more sensitive ESTA; seven of these were found to have X4 variants. All seven plus four additional specimens were classified as X4 by combined CS-PCR and 11/24/25 consensus sequencing (Tables 4 and 5).
CS-PCR detected X4-virus in a significantly greater number of specimens compared to consensus sequencing alone (p=0.024) and CS-PCR combined with consensus sequencing detected X4-virus in a significantly greater number of specimens compared to phenotypic analysis by Trofile (n=27; p<0.004). While only a small number of specimens (n=19) were phenotyped by ESTA, the difference in detection between CS-PCR combined with consensus sequencing versus ESTA was not significantly different (p=0.134) and the strength of agreement was considered moderate (kappa=0.596; 95% confidence interval 0.274, 0.918).
With ESTA designated as the gold standard, CS-PCR combined with consensus sequencing had a concordance of 79% (15/19), a sensitivity of 100% (7/7), and a specificity of 67% (8/12).
Discussion
An economical and sensitive PCR-based assay was developed to determine whether research or clinical specimens harbor HIV-1 variants that can use the CXCR4 coreceptor. Semiquantification of the input viral target ensures that the assay is sensitive to detect minor viral variants (∼1–3%). X4 genotypes are identified by combining data from a consensus sequence and CS-PCR. CS-PCR is relatively inexpensive compared to phenotypic assays, and is more sensitive compared to consensus sequencing alone.
During and following the development of CS-PCR, 60 plasma specimens from HIV-1 subtype B-infected individuals were tested. CS-PCR detected X4 variants 17% more often compared to 11/24/25 consensus sequencing alone, and in a limited comparison, 16% more often than phenotypic analysis by ESTA. The increased detection of X4 by CS-PCR compared to ESTA may be due to quantification of the input viral templates prior to CS-PCR. Input viral RNA templates are not quantified prior to testing by ESTA, and so the assay sensitivity is limited by the number of env cDNA that actually get screened for coreceptor usage. Recent massive parallel sequencing also detected minor X4 populations at a higher frequency compared to ESTA. 19 –21 Additionally, the presence of basic amino acids at codons 11, 24, or 25 does not guarantee that the variant will use CXCR4 efficiently enough to be detected using a culture-based assay. Given that HIV-1 utilization of CXCR4 evolves over time, 1,9 genotypic, but not phenotypic, assays can potentially detect viruses that have begun the evolutionary process toward use of CXCR4. CS-PCR permits the detection of these variants before they become fit, allowing one to track the evolution of low-level variants while monitoring other host variables (i.e., immune system markers).
Multiple caveats inherent to PCR constrain the use of CS-PCR. Most relevant is the high degree of variability in HIV-1 env that can lead to false-negative and false-positive classifications. Minor viral populations with mismatches or insertions/deletions near the target codons will go undetected. X4 sequences can vary extensively from the consensus and in some cases are associated with insertions close to c11 and deletions close to c25 (personal observations). A prime example in this study was sample #5, which was classified based solely on the sequence of codon 11. An insertion beyond c14 and a probable deletion at c25 made the consensus sequence indecipherable. Conversely, use of a low annealing temperature to amplify divergent sequences may allow nonspecific amplification, especially if the engineered primer mismatches happen to be complementary to the target HIV-1 sequence. A computer algorithm to screen the consensus sequence for bases found to cause false-positive CS-PCR can be written to simplify analysis.
At this juncture, CS-PCR is designed to detect the most common codons associated with CXCR4-ultizing HIV-1. Rare codons or changes at other associated sites, including those located outside the V3 loop, will not be detected. Adding reactions to screen for additional codons would increase the cost and complexity of the assay. However, it would be relatively easy to design CS-PCR for any target codon of interest —for example, monitoring the loss of N-linked glycosylation sites associated with coreceptor utilization. 22,23 An additional constraint is that CS-PCR was specifically designed for subtype B. Additional primers would be needed to test other subtypes.
CS-PCR is also limited in that it cannot quantify X4 variants. Detection of an apparent low frequency of X4-associated sequence could be because the target codon is truly at a low concentration, or because of multiple mismatches between the viral sequence and the primers. Recent studies indicate that the absolute number 24 and/or the concentration 7 of X4 variants appear to have clinical relevance. These reports note that individuals with low levels of X4 (<2% or<2.8 log copies X4/ml plasma) estimated by 454-pyrosequencing benefited from the addition of maraviroc, a CCR5 antagonist, to combination antiretroviral therapy. CS-PCR has not been evaluated to determine if it can predict clinical outcomes of treatment with CCR5 antagonists, although these studies are underway.
In summary, a PCR-based protocol to detect HIV-1 X4 variants was developed, and in preliminary analysis is more sensitive than consensus sequencing alone and is comparable to the phenotype assay ESTA. Importantly, the cost of performing CS-PCR is relatively modest by comparison to commercial phenotypic assays. Several potential weaknesses of CS-PCR are noted; most prominent is that sequence variation in the V3 loop of HIV-1 env will likely result in occasional false-negative and false-positive classifications.
Footnotes
Acknowledgments
The authors acknowledge and greatly appreciate Dr. Steven Douglas, Nancy Tustin, and their IMPAACT Immunology Specialty Laboratory (NIH IMPAACT Grant AI068632) for facilitating the transfer of samples and data from Children's Hospital of Philadelphia. Support for sample collection was provided by Grant P01 MH076388; Neurokinin-1R (SP Receptor) Antagonists for HIV-1 Therapy (Douglas, Steven D.); Project 4 (Tebas, Pablo); and Core B (Manak, Mark). We also want to acknowledge Joan Dragavon and Erin Goecker for facilitating the transfer of samples and data from the University of Washington CFAR Clinical Retrovirology Laboratory Core (AI-27757). We acknowledge and are exceedingly thankful to Dr. Richard Harrigan, Winnie Dong, Chanson Brumme, and Luke Swenson at the British Columbia Centre for Excellence in HIV/AIDS (Vancouver, Canada) and the Pfizer MOTIVATE team for providing the PCR products and 454-pyrosequencing data to optimize the ΔC t cutoffs. This work was supported by a Virology Developmental award from the International Maternal Pediatric Adolescent AIDS Clinical Trials Group (IMPAACT) (1 U01 AI068632 to LMF). The overall support for the IMPAACT was provided by the National Institute of Allergy and Infectious Diseases (NIAID) (U01 AI068632), the Eunice Kennedy Shriver National Institute of Child Health and Human Development (NICHD), and the National Institute of Mental Health (NIMH) (AI068632). The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Author Disclosure Statement
No competing financial interests.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.