
Abstract
HR212, a recombinant protein composed of the heptad repeat, is a rationally designed human immunodeficiency virus type 1 (HIV-1) fusion inhibitor. This protein can be easily produced by Escherichia coli at a low cost. Previously, studies indicated that HR212 can efficiently inhibit the entry and replication of both laboratory and clinical HIV-1 strains, and this protein is more stable and less sensitive to proteinases than T20. The procedure of HIV-1 entry into the host cells can be divided into three main steps: gp120–CD4 interactions, coreceptor binding, and gp41 six-helix bundle formation and subsequent membrane fusion. The present study demonstrates that HR212 does not block gp120–CD4 binding or interfere with binding to the coreceptors CXCR4 and CCR5. Instead, HR212 efficiently blocks the six-helix bundle formation between peptides derived from the N-terminal heptad repeat (NHR) and the C-terminal heptad repeat (CHR) region of gp41. Fluorescence native polyacrylamide gel electrophoresis (FN-PAGE) indicated that HR212 could form a complex with peptide N36 to block gp41 fusogenic core formation. These results suggest that HR212 inhibits HIV-1 entry by targeting the NHR region of gp41. Therefore, HR212 can potentially be developed as a novel, high-efficiency, specific HIV-1 entry inhibitor.
Introduction
H
Peptide T20 (also known as enfuvirtide or Fuzeon), the only FDA-approved HIV fusion inhibitor, is effective in salvage therapy for HIV/AIDS patients who have failed to respond to current antiretroviral therapeutics, including reverse transcriptase inhibitors (RTIs) and protease inhibitors (PIs). However, the emergence of T20-resistant mutants, the high production (synthesis) costs, and the high-dose treatment (200 mg/day) of T20 have greatly restricted its clinical use. 9,10 The development of new classes of drugs targeting the gp41 protein is therefore urgently needed.
Previously, we have developed two novel HIV fusion inhibitors by linking HR1 (a peptide derived from the N-terminal heptad repeat of gp41) to the C terminus of the HR1–HR2 complex (HR121) or HR2 (a peptide derived from the C-terminal heptad repeat of gp41) to the N terminus of the HR1–HR2 complex (HR212). These recombinant proteins, which are easily and cheaply produced using an Escherichia coli expression system, significantly inhibited the virus–cell fusion with IC50 values as low as nanomolar concentrations in a luciferase reporter virus system. 11 The HR212 protein was further demonstrated to be effective for inhibition of both a laboratory HIV-1 strain and clinical isolates, including T20-resistant mutants. In both cases, the protein was effective at low nanomolar concentrations and, in contrast to T20, was insensitive to proteinase K digestion. 12 NCCG-gp41 and C52L are two additional, rationally designed, bacterially expressed HIV-1 gp41 fusion inhibitors. 13,14 However, the inhibitory activity of these two compounds is not as potent as that of HR212. In this study, we focused on the potential mechanism of action of the HR212 protein and demonstrated that it did not interfere with HIV binding to the primary receptor CD4 or to the coreceptors. Instead, HR212 efficiently blocked the gp41 6-HB formation. This potent and inexpensive HIV-1 entry inhibitor may have potential utility as a therapeutic drug. Additionally, HR212 can be expressed by a probiotic organism, Lactobacillus reuteri, in the vagina to prevent HIV-1 sexual transmission, thus acting as a microbicide. 15
Materials and Methods
Cells and reagents
HIV-1IIIB chronically infected H9 cells (H9/HIV-1IIIB), MT-2 cells, U373-MAGICXCR4CEM cells, monoclonal antibody (mAb) 12G5, and mAb NC-1 were obtained from the National Institutes of Health AIDS Research and Reference Reagent Program (Bethesda, MD). HR212 protein was prepared and purified as previously reported. 11 Peptides N36, C34, C34-biotin (biotin was conjugated to the N terminus of C34), N36-F, and C34-F (FITC was added to the N terminus of N36 and C34, respectively) were synthesized and purified to homogeneity (98% purity) at GL Biochem Ltd., Shanghai, China.
Cell–cell fusion assay
HIV-1-mediated cell–cell fusion was detected as described previously.
12
A total of 6×104 MT-2 cells (50 μl) were incubated with 2×104 H9/HIV-1IIIB cells (50 μl) in 96-well tissue culture plates at 37°C in the presence or absence of HR212 or T20. Both cells were cultured in RPMI-1640 medium containing 2 mM
Inhibition of gp120 binding to CD4
The inhibitory activity of HR212 on the binding of gp120 to CD4 was detected using an enzyme-linked immunosorbent assay (ELISA) as described previously. 17,18 Briefly, 96-well polystyrene plates were coated with 100 μl of an anti-gp120 antibody (eENZYME, Gaithersburg, MD) at 2 μg/ml in 0.85 M carbonate-bicarbonate buffer (pH 9.6) at 4°C overnight. The wells were then blocked with 1% dry fat-free milk in phosphate-buffered saline (PBS) at 37°C for 1 h. Then, 100 μl of recombinant gp120 (Bal/Clade B) (eENZYME, Gaithersburg, MD) at 0.5 μg/ml was added and incubated at 37°C for 1 h, followed by three washes with PBS containing 0.05% Tween 20 (PBS-T). The HR212 (20 μM) and soluble CD4 (eENZYME, Gaithersburg, MD) at 0.25 μg/ml were added together and incubated at 37°C for 1 h. An anti-CD4 mAb (RPA-T4, 10 μg/ml) was used as a positive control. 19 After three washes, rabbit anti-sCD4 IgG (0.25 μg/ml, 100 μl/well) was added and incubated at 37°C for 1 h. Binding of rabbit anti-sCD4 IgG was determined by the sequential addition of biotinylated goat-antirabbit IgG, streptavidin-labeled horseradish peroxidase (SA-HRP) (Boster, Wuhan, China), and the substrate 3,3′,5,5′-tetramethylbenzidine (TMB) (Sigma, USA). After the reactions were terminated by the addition of 1 M H2SO4, absorbance at 450 nm (A 450) was recorded using a microplate reader (GENios, Tecan, Switzerland).
Binding of anti-CXCR4 antibody to CXCR4-expressing cells
A cell-based ELISA was performed to evaluate the HR212-mediated inhibition of mAb 12G5 binding to CXCR4-expressing cells. 20 Briefly, U373-MAGICXCR4CEM cells expressing CXCR4 molecules on the surface were seeded into a 96-well plate and cultured into a monolayer at 37°C overnight. Cells were fixed with 5% formaldehyde at room temperature for 5 min, and then washed three times with PBS-T and blocked with 5% dry fat-free milk in PBS for 1 h at 37°C. HR212, at a concentration gradient, was added into the wells that were then incubated at 37°C for 30 min. Subsequently, the anti-CXCR4 mAb 12G5 was added into the wells and incubated at 37°C for 1 h. The wells were then washed three times with PBS-T to remove the unbound antibodies. Then, biotin-labeled goat-antimouse IgG, streptavidin-labeled horseradish peroxidase, and the substrate TMB were added sequentially. There actions were terminated by the addition of 1 M aqueous H2SO4 solution. Absorbance at 450 nm was recorded in a microplate reader (GENios, Tecan, Switzerland).
Binding of gp120–CD4 complex to CCR5
The inhibition of HR212 on the binding of the gp120–CD4 complex to the coreceptor CCR5 was determined by a cell-based ELISA. 21,22 HEK293T cells were transfected by calcium phosphate precipitation with plasmid encoding CCR5. Receptor expression of transfected cells was confirmed by ELISA using anti-CCR5 antibodies. Transfected HEK293T cells were plated into a 96-well plate and cultured into a monolayer at 37°C overnight. Cells were fixed with 5% formaldehyde in PBS for 5 min at room temperature. The plate was washed three times with PBS-T and blocked with 5% dry fat-free milk in PBS for 1 h at 37°C. A 1:1 mix of soluble human CD4 (eENZYME, Gaithersburg, MD) (2 μg/ml) and HIV-1 gp120 (Bal/Clade B) (eENZYME, Gaithersburg, MD) (2 μg/ml) was incubated at room temperature for 15 min prior to its addition to the plate in the presence of HR212 (20 μM) or maraviroc (Pfizer) (1 μM). The plate was incubated at 37°C for 1 h and washed with PBS-T. Anti-gp120 antibody (eENZYME, Gaithersburg, MD) (0.5 μg/ml in PBS) was added to each well and incubated at 37°C for 1 h. The plate was then washed three times with PBS-T to remove the unbound antibodies. Then, biotin-labeled goat-antimouse IgG, streptavidin-labeled horseradish peroxidase, and the substrate TMB were added sequentially. After 5 min at room temperature, reactions were terminated by the addition of 1 M aqueous H2SO4 solution. Absorbance at 450 nm (A 450) was recorded in a microplate reader (GENios, Tecan, Switzerland).
Native polyacrylamide gel electrophoresis (N-PAGE)
Native polyacrylamide gel electrophoresis (N-PAGE) was used to detect the inhibitory activity of HR212 on the 6-HB formation between the N and C peptides as previously described. 19,23 The N36 and/or C34 peptides in PBS were incubated with or without HR212 at a concentration gradient at 37°C for 30 min (the final concentration of N36 and C34 was 40 μM). The samples were mixed with Tris-glycine native buffer (0.5 M Tris–HCl, pH 8.8, 20% glycerol, and 0.1% bromophenol blue) at a ratio of 1:1 and then loaded onto a 10×1.0 cm precast 18% Tris-glycine gel (25 μl per well). Electrophoresis was carried out with a constant voltage of 125 V at room temperature for approximately 2 h. After electrophoresis, the gel was stained with Coomassie blue and imaged.
Enzyme-linked immunosorbent assay (ELISA)
A biotin-labeled C34 (C34-biotin) was used in the ELISA for detection of the 6-HB formation. 24 Briefly, mAb NC-1 (0.8 μg/well) in 0.1 M Tris–HCl (pH 8.8) was used to coat the wells of a 96-well polystyrene plate (Greiner Bio-One, Frickenhausen, Germany) at 4°C overnight. The coating buffer was then removed and the plate was blocked with PBS containing 5% horse serum at 37°C for 2 h. N36 (25 μl, 20 μM) and HR212 (50 μl) solutions at concentration gradients in PBS were added into the precoated wells and incubated at 37°C for 30 min. Then, 25 μl of C34-biotin (20 μM in PBS) was mixed into the wells. In the control experiment, N36 was preincubated with C34-biotin at 37°C for 30 min, followed by addition of HR212 at the various concentrations. After further incubation of the mixtures for 30 min at 37°C, the plate was washed extensively with PBS-T and SA-HRP and the substrate TMB were added sequentially. Absorbance at 450 nm (A 450) was determined spectrophotometrically by a microplate reader (GENios, Tecan, Switzerland). The percentage of inhibition of the 6-HB formation by HR212 was calculated by the following equation: % inhibition=[1 – (E – N)/(P – N)]×100, where E represents the absorbance in the presence of a compound and P represents the absorbance in the absence of HR212. N corresponds to the wells where neither compound nor N36 was added. The IC50 values were calculated using the computer program Origin.
CD spectroscopy
Circular dichroism (CD) spectroscopy was performed as previously described. 18,19 Briefly, peptide N36 was incubated with HR212 or PBS at 37°C for 30 min followed by the addition of peptide C34 and incubation at 37°C for another 30 min. In the control experiment, N36 was preincubated with C34 at 37°C for 30 min before the addition of HR212. The final concentration of the peptides and HR212 was 10 μM in PBS (50 mM sodium phosphate and 150 mM NaCl, pH 7.2). The individual N36 and C34 peptides were also tested. All CD spectra were acquired on a CD spectropolarimeter (Model J-810, Jasco Inc., Japan) at room temperature using a 5.0 nm bandwidth, 0.1 nm resolution, 0.1 cm path length, 4.0 s response time, and 50 nm/min scanning speed. The spectra were corrected by the subtraction of a blank corresponding to the solvent. The α-helical content was calculated from the CD signal by dividing the mean residue ellipticity at 222 nm by the value expected for 100% helix formation (–33,000 degrees cm2 dmol–1) as described previously. 25
Fluorescence native polyacrylamide gel electrophoresis (FN-PAGE)
Fluorescence native polyacrylamide gel electrophoresis (FN-PAGE) was used to determine the interaction between HR212 and peptide N36 or C34. The FN-PAGE experiments were performed using the same conditions as the N-PAGE experiments described above, except that the N36 and C34 peptides were replaced by FITC-conjugated peptides (N36-FITC, C34-FITC). HR212 was incubated with either N36-FITC or C34-FITC in PBS (the final concentration of HR212 and peptides was 10 μM) at 37°C for 30 min before the addition of native sample buffer (0.5 M Tris–HCl, pH 8.8, 20% glycerol, and 0.1% bromophenol blue). Immediately after electrophoresis, the gel was scanned in a Typhoon 9200 image scanner (Amersham, Sweden) using a 488-nm excitation wavelength.
Results
HR212 inhibits HIV-1 entry by blocking cell–cell fusion
The fusion between HIV-infected and uninfected cells is the critical step of HIV entry into new target cells. 17 Therefore, it is essential to determine whether HR212 inhibits HIV-1-mediated cell–cell fusions. A syncytia formation assay was performed to detect the inhibitory activity of HR212 on cell–cell fusion. As shown in Fig. 1A, the fusion of HIV-1IIIB-infected H9 cells with uninfected MT-2 cells was inhibited by HR212 and T20 in a dose-dependent manner, with the IC50 values of 6.6±0.8 nM and 15.3±1.9 nM, respectively. However, zidovudine (AZT), as a reverse transcriptase inhibitor, could not inhibit cell–cell fusion. These results indicate that HR212 and T20 can act as effective inhibitors of cell–cell fusion mediated by HIV-1 infection.

HR212 does not block gp120–CD4 binding or interfere with coreceptor binding
HIV-1 entry is initiated by the binding of the viral gp120 protein to the CD4 receptor on the host cell surface; this is followed by an interaction between the gp120–CD4 complex with the coreceptor (CXCR4 or CCR5). This interaction leads to the formation of the fusion-active 6-HB of the gp41 subunit, which subsequently causes membrane fusion. To identify the role of HR212 in the inhibition of HIV-1 entry, the following experiments were performed. A CD4-based ELISA was carried out to determine whether HR212 blocks gp120–CD4 binding. As shown in Fig. 1B, an anti-CD4 mAb (RPA-T4) efficiently blocked gp120–CD4 binding (60 nM), whereas HR212 had no activity, even at concentrations of 20 μM, suggesting that HR212 does not function in blocking gp120 binding to CD4. Next, we carried out a cell-based ELISA to detect whether HR212 is capable of blocking CXCR4-mediated interactions. The CXCR4-specific mAb 12G5 and AMD3100, a potent CXCR4 antagonist, 26 were employed in these experiments, as described previously. 17 As shown in Fig. 1C, binding of 12G5 to a CXCR4-expressing cell line was significantly inhibited by AMD3100 (10 μM). In contrast, HR212 had no inhibitory activity even at concentrations of 20 μM. In another experiment, we tested whether HR212 inhibited gp120–CD4 complex binding to cells expressing CCR5. As shown in Fig. 1D, maraviroc, a CCR5 antagonist, 22 completely blocked binding of the gp120–CD4 complex to CCR5 at 1 μM, whereas HR212 had no significant inhibitory activity at 20 μM. These results indicate that HR212 does not block HIV-1 binding to either the primary receptor CD4 or the coreceptors CXCR4 and CCR5.
HR212 blocks gp41 6-HB formation
The conformational change of gp41 to form the 6-HB structure is critical for membrane fusion of HIV-1 with target cells. We examined the effect of HR212 on the formation of the 6-HB fusion complex by N-PAGE analysis. The peptides moved in the electric field according to their natural charge, shape, and size. As shown in Fig. 2A, peptide N36 (lane 1) showed no band in the gel because it carries a net positive charge. Peptide C34 (lane 2) showed a single band located near the bottom of the gel. HR212 (lane 3) showed a single band on the top of the gel. The mixture of N36 and C34 peptides (lane 4) showed two bands: the lower one located at the same position as the C34 peptide and the upper one corresponded to the size of the 6-HB formed by the N36 and C34 peptides. This was confirmed by Western blot using the mAb NC-1 (lane 8), which specifically recognizes the 6-HB structure formed by the N36 and C34 peptides but does not interact with isolated N36 or C34. 27 When N36 was preincubated with HR212 before the addition of C34 (lane 5), there was a significantly decreased intensity of the 6-HB band and an increased intensity of the C34 band, indicating that the formation of the gp41 6-HB between N36 and C34 was competitively inhibited by HR212. This was accompanied by an accumulation of free C34. However, if C34 was preincubated with HR212 followed by the addition of N36 (lane 6), the intensity of the 6-HB band was only slightly decreased. This indicates that the HR212-mediated inhibition of the formation of the gp41 6-HB depends mainly on binding with N36. Furthermore, when HR212 was added after the incubation of N36 and C34 (lane 7), the 6-HB band appeared with almost the same intensity as in lane 3. Overall, the results indicated that HR212 inhibited gp41 6-HB formation via binding with N36, but it could not function once the 6-HB had already formed. Figure 2B shows the dose-dependent inhibitory effects of HR212 on 6-HB formation by N-PAGE analysis. The overall IC50 was 5.4±1.3 μM.
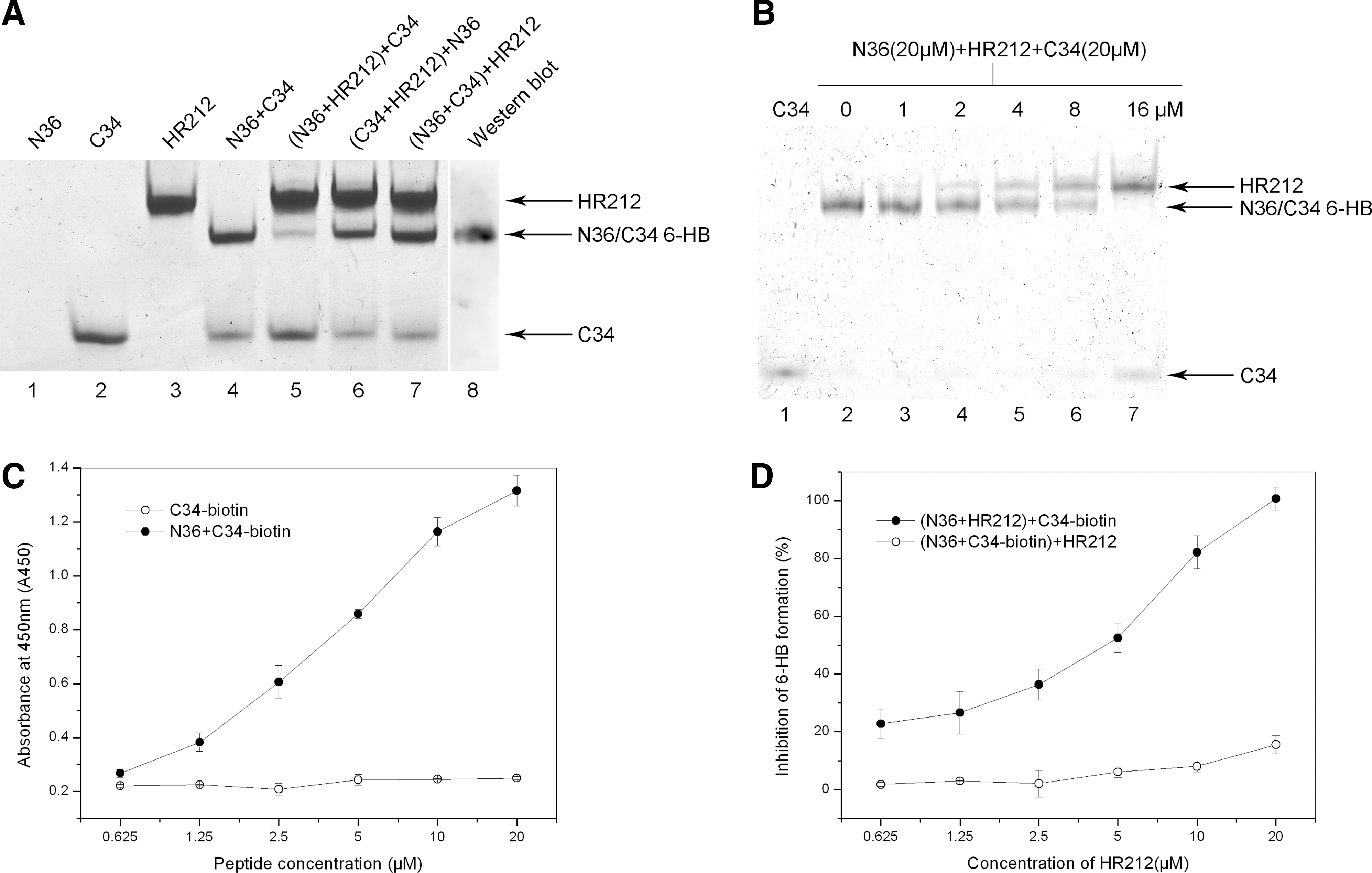
HR212 inhibited the gp41 6-HB formation as determined by native polyacrylamide gel electrophoresis (N-PAGE) and ELISA.
ELISAs were also conducted to confirm the inhibition of the 6-HB formation by HR212. Figure 2C shows that the ELISA can detect the N36/C34-biotin complex but not the individual C34-biotin because the mAb NC-1 used in the experiment is specific to the 6-HB complex. 27 Figure 2D shows that HR212 can efficiently inhibit the formation of the 6-HB complex in a dose-dependent manner with an IC50 of approximately 4.5±0.8 μM preincubated with N36 before the addition of the C34-biotin. However, if HR212 was added after the incubation of N36 and C34-biotin, the 6-HB complex formed was not disturbed by the introduction of the HR212 protein. When the final concentration of HR212 reached 20 μM (Fig. 2D), a low level inhibition of 6-HB was observed, which may be due to the capture of the 6-HB formed by HR212 by the mAb NC-1. 27 This capture may affect the interaction between NC-1 and the 6-HB formed by N36 and C34-biotin.
HR212 disrupted the α-helical structure of the gp41 6-HB complex
Previous studies demonstrated that the individual C34 or N36 peptides do not adapt any stable conformation (random coils). However, the gp41 6-HB complex formed by these two peptides shows an α-helical coiled-coil conformation. 19 Our conformational study also revealed that the mixture of the two peptides showed a saddle-shaped negative peak in the far UV region with significantly increased molar ellipticity ([θ]) at 222 nm (α-helical) compared to the individual peptide, as illustrated in the CD spectrum (Fig. 3A). Furthermore, we determined a salient α-helical character of HR212 with double minima at 208 and 222 nm in the CD spectra (Fig. 3B). However, when HR212 was added to a preincubated mixture of N36 and C34 (N36+C34+HR212), the α-helical content decreased significantly in comparison with that of the gp41 6-HB (N36+C34) and the individual HR212, suggesting that HR212 is capable of disrupting the α-helical structure of the gp41 6-HB. Additionally, when HR212 was preincubated with N36 before adding the C34 peptide (N36+HR212+C34), the α-helical content was also dramatically decreased, in accordance with the inhibition of 6-HB formation by HR212.
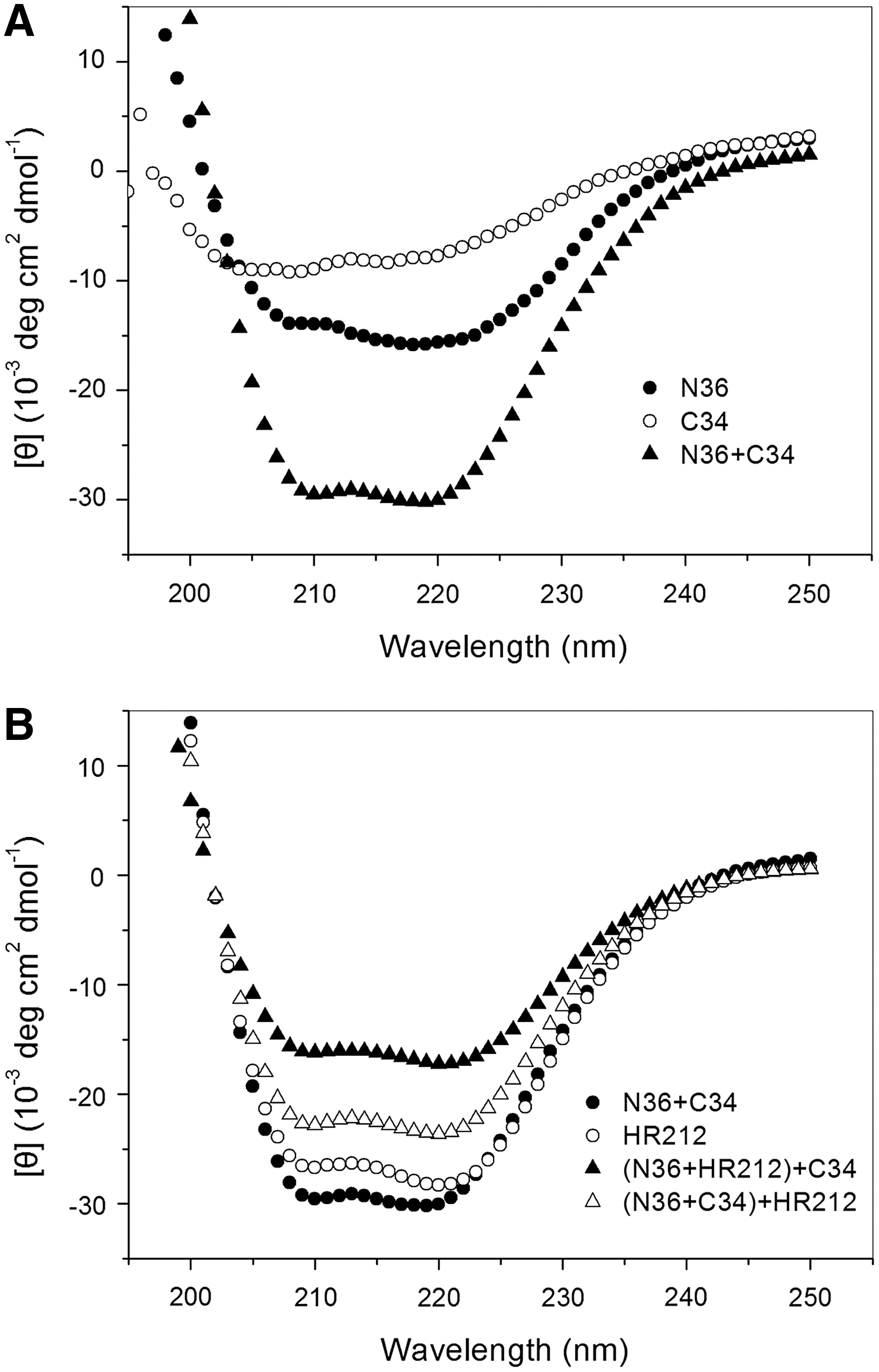
CD spectra illustrating HR212 disruption of the α-helical conformation of the complex N and C peptides.
Inhibition of 6-HB formation by HR212 is mainly correlated with the interaction between N36 and HR212
To investigate the potential mechanism of inhibition of the 6-HB formation by HR212, we used FN-PAGE to determine the interaction between HR212 and the gp41peptides (N36 and C34). As shown in Fig. 4, there was no band observed when N36-FITC was electrophoresed in the isolated form (lane 1) because the peptide N36-FITC has a net positive charge causing it to migrate upward and preventing it from entering the gel. Consequently, this peptide appeared as a faint band stacked on the top of the gel. The isolated C34-FITC peptide showed one band at the bottom of the gel (lane 2). However, if N36-FITC was mixed with C34-FITC, it appeared as one band in the middle of the gel (lane 4), which corresponded to the 6-HB complex. The isolated HR212 showed no band because it did not have a fluorescent label (lane 3). When HR212 was mixed with N36-FITC, it appeared as one band that corresponded to the N36-FITC/HR212 complex (lane 5). However, in the mixture of HR212 and C34-FITC, the only band observable was that of free C34-FITC (lane 6). These results suggest that the N36-FITC and C34-FITC peptides can form the 6-HB structure, but HR212 can interact only with the N36-FITC peptide to form a complex. There were no interactions between HR212 and C34-FITC.
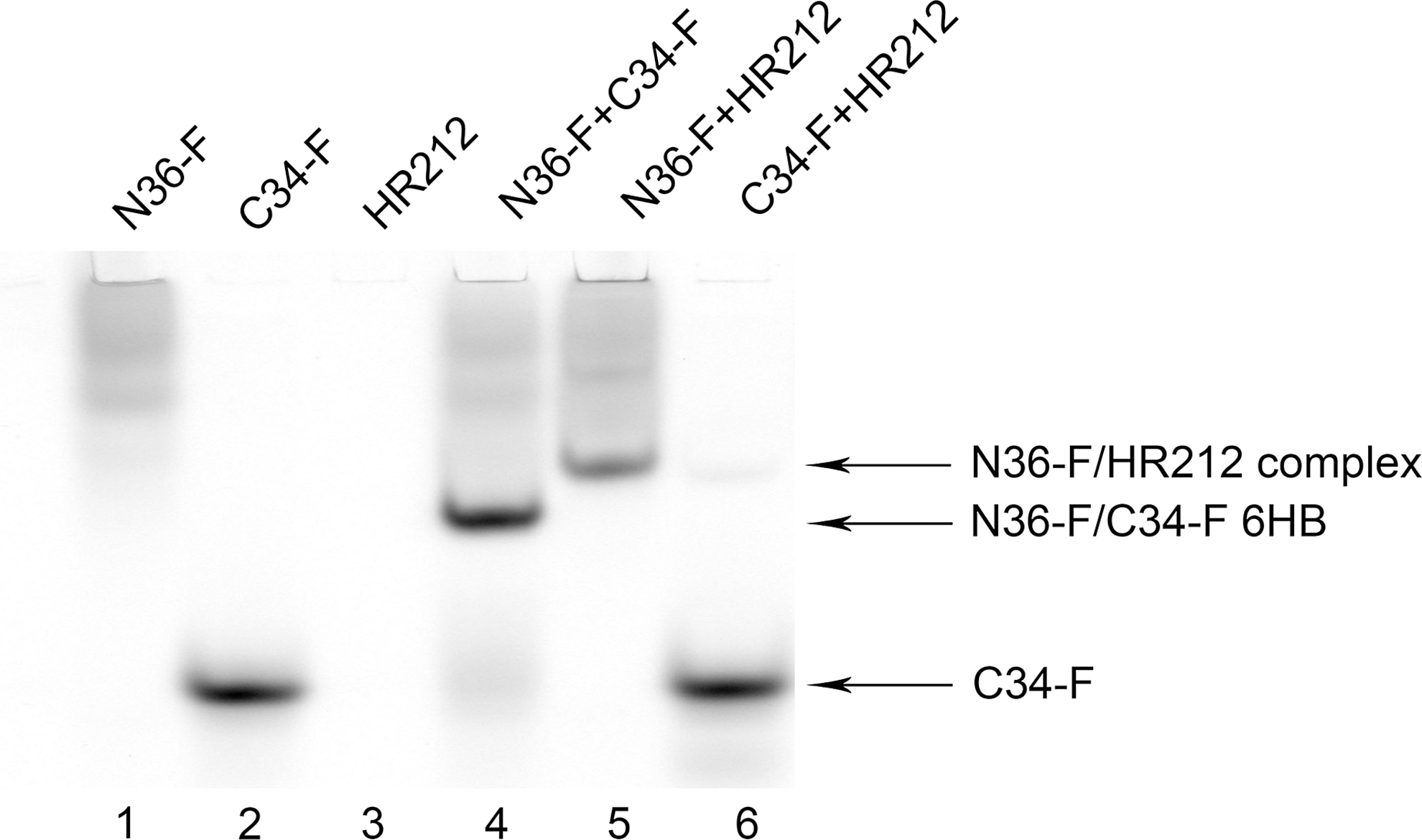
The interaction between HR212 and the N36-FITC and C34-FITC peptides was determined by FN-PAGE. The final concentrations of the peptides and HR212 were 10 μM. The gel was imaged using a Typhoon 9200 image scanner (Amersham, Sweden).
Discussion
T20, the first FDA-approved HIV fusion inhibitor, has been widely used in clinics to treat HIV-infected patients since 2003. 28 Because T20 is commercially produced by chemical synthesis involving 106 steps, the future applications are limited by the high cost of production. Like other anti-HIV drugs, T20 also has the problem of rapidly inducing drug resistance. 29 Therefore, it is essential to develop a new HIV fusion inhibitor with low cost. The recombinant protein HR212, which is efficacious against both laboratory and clinical HIV-1 strains, including T20-resistant mutants, can be easily produced by an E. coli expression system at low cost. Therefore, it is valuable to develop HR212 as a novel HIV-1 entry inhibitor.
The syncytium formation assay revealed that HR212 is capable of blocking HIV-1-mediated cell–cell fusion in the low nanomolar range, highlighting its potential as an active HIV-1 entry inhibitor. To clarify the inhibitory mechanism of HR212, we further investigated the action of HR212 in the three key steps of virus entry. We examined the effects of HR212 on viral binding to the CD4 receptor and the coreceptor and on the conformational changes in the gp41 complex. Our results indicated that HR212 does not block HIV-1 gp120 binding to CD4 or the binding to the coreceptors CXCR4 and CCR5 but it does efficiently block gp41 6-HB formation.
Peptides derived from HIV-1 gp41 CHR and NHR have been extensively studied previously. The HIV gp41 C-terminal heptad repeat contains multifunctional domains and T20 and C34, both of which were peptides derived from the HIV-1 gp41 CHR, inhibit HIV-1 entry with distinct mechanisms of action. 19,30 HR212 is a recombinant protein that contains one molecule of HR1 and two molecules of HR2. 11 We have previously demonstrated that three molecules of the HR212 protein could form a stable 6-HB with three free heptad-repeats (HR2) exposed. This structure allows efficient binding to the N-peptide (N36)-derived NHR (HR1), thus disrupting gp41 6-HB formation. Because HR212 contains one molecule of N-peptide (N34) derived from the gp41 NHR (HR1) region, it can interact with the C34 peptide. As most of HR1 in HR212 is already bound to the HR2 region to form the 6-HB with very few hydrophobic grooves exposed, the interaction between HR212 and the C34 peptide is nearly negligible. The FN-PAGE results also indicated that N36 can bind with HR212 to form a stable complex, but C34 cannot. CD spectroscopy of HR212 suggested that HR212 could self-associate to form a coiled coil structure with high levels of α-helical content under physiological conditions. This structure in a peptide-based fusion inhibitor is a key factor for the inhibitory activity of HR212 because it could reinforce the binding affinity to the NHR region. 31 Additionally, this thermostable structure may also result in decreased metabolism by proteases and improved pharmacokinetics in vivo. 32
A recent study indicated that HIV predominantly used cellular endosomes as transport carriers to gain access to the cytoplasm and membrane fusion preferentially occurred at the endosomes. 33 This would elicit protection to HIV from fusion inhibitors, as the surface exposition of the gp41 extended conformation is reduced. Hence, the capability of peptide fusion inhibitors to partition to the membrane and remain there while the virus is internalized, together with a cell membrane patch, can prove to be a decisive factor concerning their inclusion on the endosome, leading to increased antiretroviral efficacy and potency. Further modification to HR212 (such as being linked with a lipid) may improve the antiviral activity.
Recently a novel therapeutic approach has been developed to combat HIV sexual transmission in women. This approach uses the probiotic organism Lactobacillus reuteri, which safely colonizes the human vagina and prevents microbial infections, and engineers it to produce anti-HIV proteins. 15 A study in female rhesus macaques showed that the use of vaginally delivered small molecule and HIV peptide inhibitors effectively protected the macaques against vaginal SHIV challenges. 34 Previous studies indicated that HR212 can significantly inhibit entry and replication of laboratory and clinical HIV-1 strains, and compared to T20, HR212 is more stable in cell culture supernatants and less sensitive to proteinases. 12 These results imply that HR212 can be used in this new therapeutic approach. The HR212 sequence can be stably inserted into the chromosome of Lactobacillus reuteri, allowing HR212 to be persistently expressed in the vagina, and thus blocking HIV entry.
Footnotes
Acknowledgments
We are grateful to Dr. Paul Chu, a visiting professor at the Institute of Microbiology Chinese Academy of Science, and Dr. Xuebin Li of the CAS Key Laboratory of Pathogenic Microbiology and Immunology, Institute of Microbiology, Chinese Academy of Sciences, for critical reading of the manuscript. We thank the NIH AIDS Research and Reference Reagent Program for providing the cell lines and antibodies. We are grateful to Shuwen Liu at Southern Medical University, Guangzhou, China for excellent technical assistance. This work was supported by grants from the Major National Science and Technology Specific Projects of China (nos. 2009ZX09103-747, 2009ZX10004-305, and 2009ZX09301-014-1) and the National Natural Science Foundation of China (NSFC, grant 30900758).
Author Disclosure Statement
No competing financial interests exist.