
Abstract
Herpes simplex virus-2 (HSV-2) suppression with acyclovir or valacyclovir reduces HIV-1 viral RNA levels; one hypothesis is that HSV-2 suppression reduces immune activation. We measured T cell immune activation markers among women participating in a randomized placebo-controlled trial of valacyclovir to reduce HIV-1 RNA levels among pregnant women. Although valacyclovir was associated with lower HIV-1 RNA levels, the distribution of both CD4+ and CD8+ CD38+HLA-DR+ T cells was not different among women taking valacyclovir when compared to women taking placebo. Further study is needed to understand the mechanism of HIV-1 RNA reduction following herpes suppression among those coinfected with HIV-1 and HSV-2.
Introduction
C
Materials and Methods
This study was nested within a randomized, double-blind clinical trial, evaluating the efficacy of 500 mg valacyclovir twice daily or matching placebo for reducing maternal HIV-1 RNA levels in plasma, genital secretions, and breast milk, as described by Drake et al. 8 From 2007 to 2009, pregnant, HIV-1-seropositive, HSV-2-seropositive women seeking antenatal care were recruited and followed at a public health clinic in Nairobi, Kenya. Eligible women were ≥18 years of age, WHO stage 1 or 2, with a CD4 count >250 cells/μl. HIV-1-seronegative nonpregnant female subjects ≥18 years of age were recruited at voluntary counseling and testing centers in Nairobi from 2009 to 2010 as controls for a different cohort. Both study protocols were approved by the Kenyatta National Hospital Ethical Review Committee and the University of Washington Institutional Review Board. The clinical trial was registered (ClinicalTrials.gov, identifier NCT 00530777).
At 34 weeks gestation, participants provided written informed consent and were randomized; study drugs were taken from 34 weeks until 1 year postpartum. In addition to study drug, mothers received short-course prevention of mother-to-child (PMTCT) antiretroviral (ARV) regimens according to Kenyan national guidelines. Blood specimens were collected at all study visits; CD4 counts and HIV-1 RNA levels were determined at enrollment, 6 months and 12 months postpartum. T cell immune activation markers were measured at 6 and 12 months postpartum. HIV-1-seronegative female participants in the control population gave written consent, were enrolled, and gave a blood specimen at one time point only.
Maternal HIV-1 serostatus was determined with rapid-testing protocols and confirmed using enzyme-linked immunosorbent assay (ELISA) (Vironostika HIV Uni-Form II, bioMérieux). HSV-2 serostatus was determined using HerpeSelect ELISA (Focus Technologies) with positive result defined as an index value ≥3.5. HIV-1 RNA assays were performed using a transcription-mediated amplification method validated for Kenyan HIV-1 subtypes (Gen-Probe). Immune activation markers were measured on fresh whole blood specimens; specimens were surface-stained with fluorochrome-conjugated antibodies, and each specimen was run with isotype controls on a 4-color flow cytometer (FACSCalibur, BectonDickinson). The following two premixed staining combinations were used: anti-CD4-FITC/anti-CD38-PE/anti-CD3-PerCP/anti-HLA-DR-APC, and anti-CD8-FITC/anti-CD38-PE/anti-CD3-PerCP/anti-HLA-DR-APC (BectonDickinson). Single-stain controls and unstained controls were run daily. Flow cytometry results were interpreted using FlowJo and a logical gating strategy was used to define activated cells.
Baseline characteristics (including CD4 counts and HIV-1 RNA levels) of women in each study arm with immune activation results were compared by chi-square and Wilcoxon rank-sum tests. Distributions of activated cells, defined as CD38+, HLA-DR+ and combined CD38+HLA-DR+, in subsets of CD4+ and CD8+ T cells were compared between randomization arms using nonparametric tests. The same analyses were done to compare T cell subsets between HIV-1-seropositive participants and HIV-1-seronegative controls. Analyses were done using Stata 10.
Results
Of the 148 women enrolled in this study, 12 (8%) were lost to follow-up and 19 (13%) came to the clinic when the laboratory was closed; 117 women (79%) had at least one immune activation result and are described in this analysis. The women had a median age of 25, and most had one prior pregnancy, primary school education or less, and lived in poverty (Table 1). Consistent with enrollment criteria, all women were WHO stage 1 or 2, and 89% never had an HIV-1-related illness. At 6 months postpartum, 51% of study women were breastfeeding; by 12 months postpartum, 13% of study women were breastfeeding. Two women initiated ART by 12 months postpartum. Control women (n=31) had similar median numbers of sexual partners and children, but had more education and were more likely to be employed. Only eight (27%) of the control women were HSV-2 seropositive, compared to 100% of the study women, and control women were less likely to ever have an STI. Four (13%) control women were breastfeeding.
n=58.
GUD, genital ulcer disease; STI, sexually transmitted infection.
When compared to the main study results, which reported a relative reduction of HIV-1 RNA of 0.53 log10 copies/ml at 1 year postpartum among women randomized to valacyclovir, 8 the subset of women in this nested study randomized to valacyclovir also had significant relative reductions in plasma HIV-1 RNA levels of 0.6 log10 copies/ml at 6 months postpartum and 0.3 log10 copies/ml at 12 months postpartum. Only seven (12%) women taking valacyclovir and 11 (19%) women taking placebo had a prior history of genital ulcer disease (GUD). During follow-up, illness was common in both study arms: five (4%) women were treated for tuberculosis (two valacyclovir, three placebo), three (3%) women were treated for herpes zoster (one valacyclovir, two placebo), 18 (15%) received treatment for malaria (five valacyclovir, 13 placebo), and 23 (20%) received treatment for diarrhea or helminths (14 valacyclovir, nine placebo). Three women died prior to 6 months postpartum and are not included in this analysis. Monitoring showed high adherence (>85%) to study drugs as previously reported. 9
Immune activation did not differ between women randomized to valacyclovir and women taking placebo. At 6 months postpartum, the median percentage of CD38+HLA-DR+ CD4+ T cells was 2.0% in the valacyclovir arm vs. 2.0% in the placebo arm (p=0.75); at 12 months postpartum, the median percent CD38+HLA-DR+ CD4+ T cells was 1.7% (p=0.98) in both study arms. Similarly, at 6 months postpartum, the median percentage of CD38+HLA-DR+ CD8+ T cells was 4.9% among women taking valacyclovir, compared to 5.0% among women taking placebo (p=0.95), and at 12 months postpartum, there was again no significant difference between study arms: 4.8% of cells were activated among women taking valacyclovir, compared to 4.5% among women taking placebo (p=0.72). At both 6 and 12 months postpartum, percentages of CD38 and HLA-DR activated T cells were uniformly higher among HIV-1-seropositive women in the study when compared to HIV-1-seronegative female controls (Fig. 1).
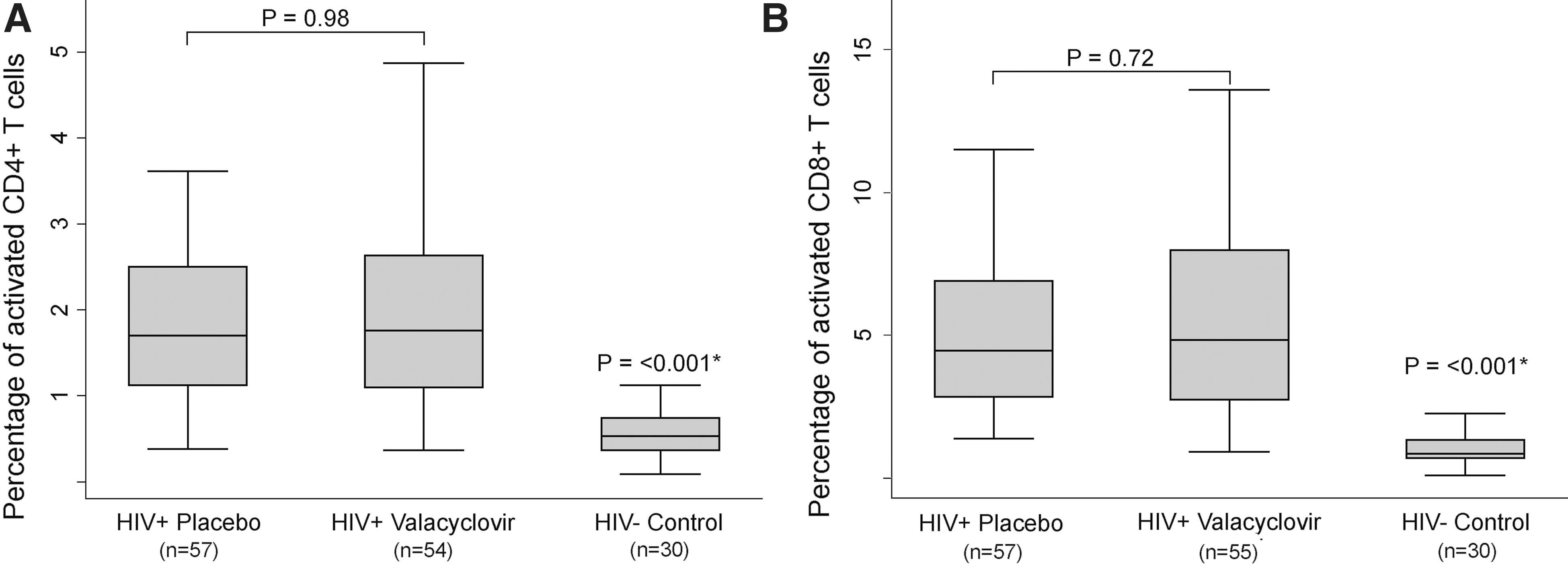
Univariate comparison of T cell activation markers. T cell activation in human immunodeficiency virus-1 (HIV-1)-seropositive study participants randomized to valacyclovir or placebo and HIV-1-seronegative Kenyan women. This figure describes activated CD38+HLA-DR+ CD4+ T cells
Discussion
We did not see any differences in immune activation between the two study arms, although women in the valacyclovir arm had significantly lower HIV-1 plasma RNA levels. This result differs from studies of other coinfections, including tuberculosis, helminths, and malaria, which have shown reductions in both HIV-1 RNA levels and systemic immune activation after treatment. 10,11 HIV-1-seropositive persons with HSV-2 who take acyclovir or valacyclovir in clinical trials have sustained decreases in HIV-1 RNA levels and reduced disease progression; our results suggest that these findings may not be related to reduced immune activation.
If acyclovir therapy does not reduce systemic immune activation, then how does it reduce HIV-1 RNA levels? There is evidence that valacyclovir interacts directly with HIV-1 reverse transcriptase if phosphorylated by human herpesviruses in tissues, 12 although it remains unclear why no acyclovir resistance develops in vivo; 13 valacyclovir may also suppress HIV-1 replication indirectly by reducing HSV-2, causing a cascade of effects including a reduction in target cells for HIV-1 replication and less HSV-2 upregulation of HIV-1 transcription factors. 14
The lack of effect of valacyclovir on immune activation that we observed may be unique to HSV-2 when compared to other coinfections. The long duration of inflammation associated with HSV-2 lesions 15 and the steady shedding of HSV-2 even among people who are asymptomatic 16 or being prophylactically treated 17 suggest that anti-herpes medications are not optimally effective in the mucosal sites where the immune activation is potentially generated. This could partly explain the failure of acyclovir to protect against HIV-1 transmission and acquisition in large randomized trials. 18,19
Our results contrast with a recent trial of valganciclovir in persons with HIV-1 and cytomegalovirus (CMV) coinfection, which showed reductions in immune activation markers among those with poor CD4 response to ART who completed 8 weeks of CMV suppression. 20 CMV-specific cells dominate the circulating memory T cell repertoire 21 and results from these two studies may signal that CMV may be a more important coinfection than HSV-2 in amplifying systemic immune activation. In addition, the HIV-1/CMV coinfected subjects were on ART, which could make the immune activation more easily downregulated.
The strengths of our study are randomization to reduce confounding, prolonged (>12 months) follow-up of participants, excellent adherence to valacyclovir, a large sample with adequate power to detect clinically significant differences in immune activation, and inclusion of an appropriate local control population. The weaknesses include lack of pre-valacyclovir measures of immune activation and unpredictable effects of recent pregnancy and breastfeeding on immune activation, although these are mitigated by the randomized trial design. Maternal illness is a confounder that could influence immune activation, but appears to be evenly distributed across study arms. We measured only two commonly cited markers of T cell activation, but other markers might reveal different results. This cohort of postpartum women with CD4 >250 cells/μl was relatively healthy, and our results may not be generalizable to other populations, or populations with lower CD4 counts.
In conclusion, 500 mg twice daily valacyclovir taken over 12 months in a group of asymptomatic HIV-1/HSV-2 coinfected African women failed to reduce systemic immune activation markers. Valacyclovir's role in controlling HIV-1 is promising, and new drugs are in development to exploit this potential. 22 Further research should be pursued to clarify how anti-herpes medications improve outcomes in HIV-1/HSV-2 coinfected individuals.
Footnotes
Acknowledgments
We gratefully acknowledge the women who participated in this study, our study staff, and the staff of Mathare North Health Centre in Nairobi. We especially thank our laboratory staff, John Gatimu and Daniel N. Matemo.
This work was supported by US National Institutes of Health (NIH) research grants (R03 HD 057773, R03 HD 057773-02S1, R01 AI076105, K24 AI087399, R01 AI06843-05 to C.F., K24 HD054314 to G.J.S., K24 AI071113 and PO1 AI30731 to A.W.); the University of Washington Center for AIDS Research (CFAR) (P30 AI027757), a Puget Sound Partners for Global Health Research and Technology Grant to B.A.R., and University of Washington Royalty Research Fund Grant #4027. GlaxoSmithKline donated study drug and matched placebo, but had no other role in the study. A.L.D. was supported by an NIH-funded University of Washington CFAR Training Grant (T32AI07140-32). A.C.R. and A.Y.L. were scholars in the International AIDS Research and Training Program, funded by the Fogarty International Center, NIH (D43-TW000007). A.C.R. was a Fogarty International Clinical Research Fellow funded by the Fogarty International Center, NIH (R24 TW007988).
Author Disclosure Statement
Dr. Wald has received grant support from GlaxoSmithKline and Antigenics and has consulted for AiCuris and Agenus. All other authors report no disclosures.