
Abstract
HIV binds specifically to the human mannose receptor (hMR) on vaginal epithelial cells that are devoid of a conventional CD4 receptor. HIV binding to hMR on vaginal epithelial cells induces the production of matrix metalloproteinase 9 (MMP9) leading to degradation of the extracellular matrix, which may increase the risk of HIV entry into vaginal epithelial cells and further transmission into distal cells. Immunofluorescent localization of hMR on vaginal epithelial cells of seronegative females from the general population included the control group (n=52) and seronegative females from serodiscordant couples. There was PCR amplification of DNA from peripheral blood mononuclear cells (PBMCs) of the serodiscordant females for the CCR5 gene flanking the CCR5-Δ32 region; PCR amplification and sequencing of the C2-V3 region of HIV variants in PBMCs and sperm of the infected male partners of the serodiscordant couples; and the presence of hMR on 0–11% of the vaginal epithelial cells of seronegative females (n=39) from serodiscordant couples and 90–95% that of a control group of females (n=52). Nine of these serodiscordant females did not show a CCR5-Δ32 deletion. The translated amino acid sequence of the C2-V3 region of the env gene of HIV-1C in PBMCs (n=9) and sperm (n=5) of the male partners showed the presence of distinct variants and the variation in PBMCs and sperm of serodiscordant males was almost similar to that of infected males from concordant couples. The presence of hMR in a smaller number of vaginal epithelial cells of serodiscordant females prevented binding and HIV entry into these cells and therefore prevented sexual transmission of HIV.
Introduction
T
The lower female genital tract is lined by stratified squamous epithelial cells (vagina and ectocervix) or a single epithelial monolayer (endocervix). It is unclear how HIV-1 particles or cell-associated virus penetrate intact mucosa and initiate infection. Studies also demonstrated that the intact vaginal epithelium has limited permeability and is refractory to the ability of molecules above 30 nm in diameter to trespass into the lower layers. 5 In addition, although the HIV virion is about ∼80–100 nm in size, the presence of HIV particles has been identified in the subepithelial cells 6 –8 and also between cells of the stratified epithelium. 9 This suggests the possibility that the virus would first bind and enter into the epithelial cells, which make them more permeable for viral entry into the subepithelial layers. However, due to lack of a conventional CD4 receptor on vaginal epithelial cells, the precise mechanism of HIV binding and entry into host cells in the reproductive tract is not known.
Recently by immunofluorescent localization, the Western blot analysis of vaginal epithelial cell lysate and reverse transcriptase polymerase chain reaction (RT-PCR) demonstrated the presence of the human mannose receptor (hMR) on vaginal epithelial cells. 10 Furthermore, HIV-1 gp120 has been shown to bind specifically to hMR and induces the production of MMP9 10 ; HIV has also been shown to bind hMR on human astrocytes and to activate MMPs, which induce degradation of extracellular matrix proteins. 11 Additionally, HIV has also been demonstrated to bind primary genital epithelial cells, to inhibit the expression of tight junction proteins, and to increase the leakiness of the epithelial layer toward HIV. 12,13 Induction of MMPs in response to HIV gp120 may therefore lead to degradation of tight junction proteins and the extracellular matrix proteins in the vaginal epithelium and basement membrane, leading to weakening of the epithelial barrier, thereby facilitating transport of HIV across the vaginal epithelium. This suggests HIV binding to hMR and entry into vaginal epithelial cells may be responsible for further transmission to distal cells and therefore may possibly increase the risk of male-to-female sexual transmission of HIV. 14 Additionally, male factors such as seminal viral load, incidence of STDs, and affinity of HIV variants from urogenital cells and secretions to host cells may also influence male-to-female sexual transmission of HIV.
Despite the high risk of sexual transmission of HIV, it is still not understood why some individuals remain uninfected even after unprotected sex with their infected sexual partner. This article describes the association of sexual transmission of HIV with hMR in vaginal epithelial cells, CCR5-Δ32 deletion, and the HIV variants in sperm and peripheral blood mononuclear cells (PBMCs) of the infected male partner.
Materials and Methods
Recruitment of study participants
Thirty-nine seronegative normal healthy females of HIV-infected male sexual partners from discordant couples were recruited. The HIV-infected male partners from these serodiscordant couples recruited for the study were found to be infected for a duration of about 3 to 7 years. The females from this group were counseled for protected sex. However, these females were not found to be consistent in engaging in protected sex, which is evident from their clinical history as well as the fact that in some cases pregnancy occurred. Nine out of 39 HIV-1C-infected male sexual partners of these seronegative females were also recruited. The second group of 52 seronegative normal healthy females of HIV-negative male sexual partners were recruited as a control group. Another group of 39 HIV-infected females of HIV-infected sexual partners (concordant couples) was also recruited for the study. All the participants were recruited from the ART Center, JJ group of hospitals, Mumbai with approval from the Institutional Clinical Ethics Committee; an informed consent form was obtained from all of them. All the study participants who were recruited demonstrated an absence of ulcerative lesions/pathological discharge in the urogenital tract and were between 20 and 45 years of age. The blood samples of infected males were collected in EDTA-coated vacutainers. A blood CD4 count was estimated by flow cytometry using a Guava Technologies cocktail of antibodies to CD4/CD3 as described earlier. 15 The semen samples of the HIV-infected male participants were collected by masturbation into a sterile polypropylene bottle after 2–3 days of ejaculatory abstinence. Total nucleic acid from blood and seminal plasma was isolated using the MagNa pure Compact Nucleic Acid Automated System (Roche Diagnostic, Germany) and viral load was estimated using Cobas TaqMan Real Time PCR (Roche Molecular Systems, Piscataway, NJ) according to the manufacturer's instructions as previously described. 15 The PBMCs from blood and sperm from semen samples were separated by the method described earlier 15 and DNA from PBMCs and sperm was isolated as mentioned below.
Isolation DNA from PBMCs
DNA from PBMCs of infected individuals was isolated using a Qiagen DNA mini kit according to the method described by the manufacturer. In brief, about 5×106 lymphocytes in 200 μl phosphate-buffered saline (PBS) were incubated at 56°C for 10 min in the presence of 20 μl QIAGEN Protease and 200 μl Buffer AL. Following centrifugation 200 μl ethanol (96–100%) was added to the pellet and centrifuged at 6000×g (8,000 rpm) using a QIAamp spin column. The filtrate was discarded and the pellet washed with 500 μl Buffer AW2 by spinning at 20,000×g (14,000 rpm) for 3 min. The pellet was then suspended in 200 μl of buffer AE and incubated at room temperature for 1 min. Following centrifugation at 6,000×g for 1 min the supernatant was estimated for DNA content at OD 260 nm and 280 nm.
Isolation DNA from sperm
DNA from sperm was isolated using a HiPurA sperm genomic DNA purification spin kit according to the manufacturer's instructions. In brief, sperm samples were washed with 10 ml and subsequently with 1 ml of Semen Wash Buffer (SEW). The sperm pellet thus obtained was suspended in 100 μl of PBS and 100 μl of Sperm Lysis Buffer (SL) and incubated at 55°C for 1.5–2 h. Following incubation with 20 μl of RNase A solution for 2 min at room temperature, 200 μl of ethanol (96–100%) was added and loaded on the HiElute Miniprep spin column and centrifuged at ≥6,500×g (≈10,000 rpm) for 1 min. The suspension was then washed twice with Wash Solution (WS) and subsequently twice with 100 μl of Elution Buffer. The DNA content was then estimated at 260 nm and 280 nm OD.
Vaginal swabs were collected from all the females recruited and evaluated for immunofluorescent localization of hMR using FITC-labeled monoclonal antibodies to hMR. PBMCs of the female partner were also investigated for CCR5-Δ32 deletion.
Immunofluorescent localization of hMR on vaginal epithelial cells
Immunofluorescent localization of hMR on vaginal epithelial cells was investigated using the method described earlier. 10 –14 In brief, vaginal smears obtained from the HIV-negative female partner of the serodiscordant couples, HIV-infected females from the concordant couples, and normal females from the control group were fixed with ice cold acetone for 20 min. The slides were washed three times with 10 mM PBS, pH 7.4, and incubated in the presence of 1% nonfat dried milk for 1 h at room temperature followed by overnight incubation at 4°C with FITC-labeled monoclonal antibody to hMR (FITC Ab-hMR) or FITC-labeled IgG (IgG FITC) (BD Pharmingen) as a negative control. Following washing three times with PBS the slides were counterstained using propidium iodide and mounted in Vectashield (Vector Laboratories, Cambridge Shire, United Kingdom). The smears were observed under a confocal microscope and the fluorescent localization was examined for 200 cells and expressed in percentage cells positive for hMR localization.
PCR amplification of CCR5 gene from PBMCs of seronegative females
The CCR5-Δ32 region of the CCR5 gene was PCR amplified using the method described earlier. 16 In brief, about 0.5–1 μg of DNA isolated from PBMCs of nine HIV-serodiscordant females was PCR amplified using a set of forward primer: 5′ ACC AGA TCT CAA AAA GAA 3′ and reverse primer: 5′ CAT GAT GGT GAA GAT AAG CCT CA 3′ (GenBank Accession number: AF009962). PCR amplification was performed using 20 mM Tris–HCl, pH 8.4, 50 mM KCl, 1.5 mM MgCl2, 200 mM dNTP, and 1.25 units of Taq polymerase. The PCR conditions were denaturation at 94°C, 5 min, followed by 35 cycles of 1 min at 94°C, 1 min at 58°C, and 1 min at 72°C, and a 10 min elongation at 72°C; PCR products were analyzed by electrophoresis using 10% acrylamide gel electrophoresis.
Genotypic characterization of HIV variants
The C2-V3 region of the HIV-1C env gene from proviral DNA in PBMCs and spermatozoa of the infected male partner of the serodiscordant couples was amplified by nested PCR as described earlier. 15,17 In brief, 0.5–1 μg of the DNA template from PBMCs and spermatozoa was PCR amplified using as a first round 20 pmol each of the ED5 and ED12 primer set in the presence of 1.25 mM MgCl, 2.5 mM dNTPs, and 2.5 units of Taq polymerase (Taq polymerase) in a total volume of 25 μl. The PCR conditions were 94°C 15 min; 3 cycles of 1 min each at 94°C, 50°C, and 72°C; 35 cycles at 94°C for 15 s, 55°C for 45 s, 72°C for 1 min, and a final extension at 72°C for 5 min. Subsequently the C2-V3 region from 5 μl of first round of product was similarly amplified by second round PCR using an ED31/ED33 set of primers. The second round PCR product was purified and sequenced by an automated DNA sequencing system.
Results
Nine out of 39 seronegative females and their HIV-1C-infected male partners provided blood samples and five of these males also provided semen samples. Blood and/or semen samples from the remaining participants could not be collected due to lack of interest in providing the samples. Vaginal swabs obtained from all the 39 seronegative females were investigated for localization of hMR in vaginal epithelial cells. The viral load estimated in blood and seminal plasma did not show a correlation (Table 1). For one of the male participants the viral load in blood was found to be undetectable but the semen viral load was 1,236 copies/ml (Table 1). Seven of these serodiscordant couples were found to have children aged 1 to 12 years who were also seronegative. Two of these couples did not provide the status about their children (Table 1).
Participant declined to provide sample.
SDC, serodiscordant couples; ART, antiretroviral therapy; hMR, human mannose receptor; SNF, seronegative females; Undet, undetectable; NE, not estimated; S-A, status not available; ZDV, zidovudine; 3TC, lamivudine; NVP, nevirapine; d4T, stavudine; EFV, efavirenz.
Expression of human mannose receptor on vaginal epithelial cells
The presence of hMR has been demonstrated by us on sperm and vaginal epithelial cells to which HIV specifically binds. 10,14 Using FITC Ab-hMR showed the specific localization of hMR on vaginal epithelial cells. The confocal images were observed in three channels: the green fluorescence color shows the presence of hMR on vaginal epithelial cells (Figs. 1a, 2a, 3a, and 4a), a green and red color shows a merged image of localization of hMR along with the nucleus (Figs. 1b, 2b, 3b, and 4b), and only the red color is seen for nuclear images (Figs. 1c, 2c, 3c, and 4c). Figure 1 shows the representative image of the localization of hMR on the vaginal epithelial cells of the seronegative females from the control group. All the cells counterstained with propidium iodide show a red-colored nucleolus and also FITC Ab-hMR localization with green fluorescence suggesting localization of hMR on all the vaginal epithelial cells (Fig. 1).
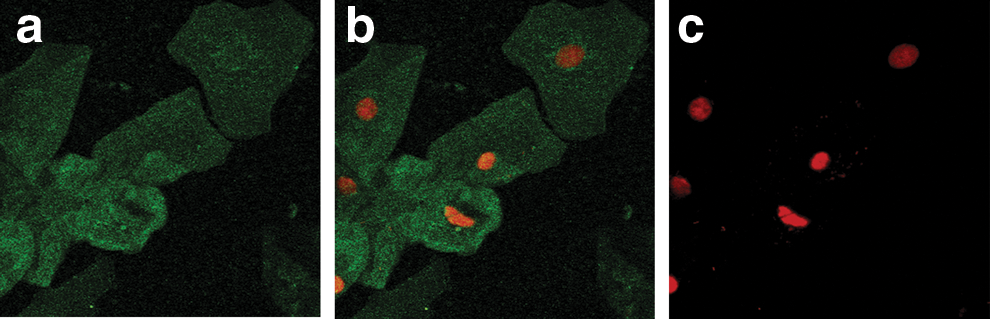
Localization of the human mannose receptor (hMR) on vaginal epithelial cells of the normal HIV. Seronegative females are from the general population using FITC-labeled monoclonal antibodies to hMR (FITC Ab-hMR). The green fluorescence shows the hMR localization on the cells. The red color shows the nuclear staining.
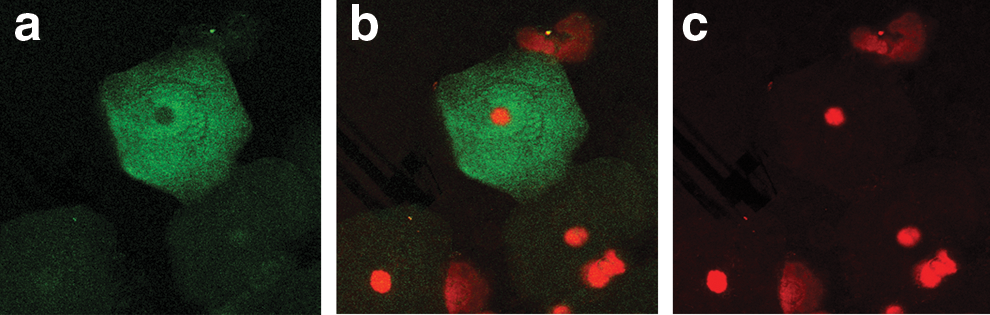
Localization of hMR on the vaginal epithelial cells of the HIV-seronegative females from serodiscordant couples. The green fluorescence shows the hMR localization on the cells. The red color shows the nuclear staining.
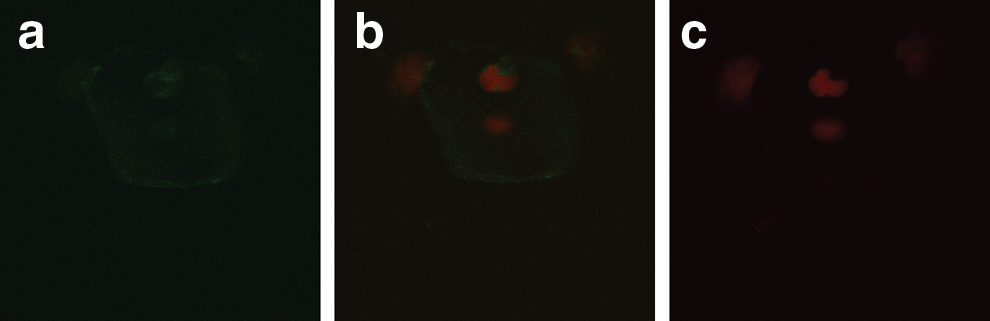
Localization of hMR on vaginal epithelial cells of HIV-infected females. Seropositive females are from general population using FITC-labeled monoclonal antibodies to hMR (FITC Ab-hMR). The green fluorescence shows the hMR localization on the cells. The red color shows the nuclear staining.
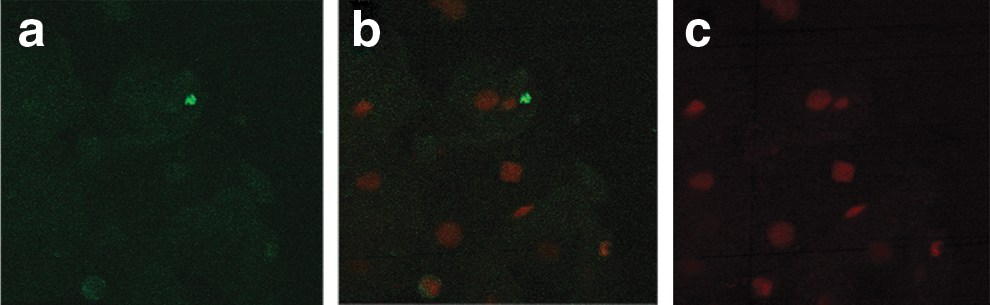
Negative control. Vaginal epithelial cells incubated with FITC-labeled IgG.
Figure 2 shows the representative image of immunofluorescent localization of hMR on vaginal epithelial cells of the seronegative females from the serodiscordant group. Cells counterstained with propidium iodide did not show green fluorescence in all the cells, suggesting the localization of hMR on the lower number of cells. Figure 3 shows the representative image of immunofluorescent localization of hMR on a lower number of vaginal epithelial cells of the HIV-infected females. Figure 4 shows that the localization of hMR on vaginal epithelial cells (negative control) using IgG FITC did not show the green fluorescence and shows only red color nuclear staining suggesting the specificity of the localization of hMR. Localization of hMR was observed on 90–95% of vaginal epithelial cells of females from the control group (n=52) from the general population (Table 2). Of the vaginal epithelial cells of 39 seronegative females from serodiscordant couples 0–11% showed the presence of hMR (Table 2). Vaginal epithelial cells of 14 seronegative females (36%) showed the complete absence of hMR (Table 2) while 10 females (25%) showed hMR localization on one to two cells and the remaining 15 females showed the localization in the range of 3–11% of the vaginal epithelial cells (Table 2). This shows that the localization of hMR in less than 11% of the vaginal epithelial cells of these seronegative females prevented HIV binding to hMR on vaginal epithelial cells. Localization of hMR was observed in 8–10% of the vaginal epithelial cells of the HIV-infected females (n=39).
90–95% of vaginal epithelial cells of the normal females (n=52) from the general population showed the localization of hMR.
8–10% of vaginal epithelial cells of the HIV-infected females showed the localization of hMR.
Analysis of coreceptor usages
The individuals with homozygous deletion of the CCR5-Δ32 gene are known to be resistant to HIV infection. Nine of these serodiscordant females who provided the blood samples were also investigated for the CCR5-Δ32 deletion. DNA from PBMCs of these nine females was PCR amplified using a pair of specific primers flanking for the CCR5-Δ32 deletion. The CCR5 wild-type homozygous genotype is of 225 bp, the heterozygous deletion of genotype shows bands of 193 bp and 225 bp, and the homozygous deletion is of 193 bp in size. The PCR amplification of DNA for the CCR5-Δ32 gene of these nine serodiscordant females showed a single band of 225 bp (Fig. 5), suggesting the presence of a homozygous wild-type CCR5 gene; therefore prevention of HIV transmission is not associated with the deletion of the CCR5-Δ32 gene.

Screening of the CCR5-Δ32 deletion in seronegative females. PCR amplification of the CCR5 gene using specific primers flanking for the CCR5-Δ32 region. No. 8: lane represents the 100-bp ladder.
Genotypic characterization of the C2-V3 region of the env gene of HIV-1C in PBMCs and spermatozoa
The C2-V3 region of the env gene of HIV-1C isolated from PBMCs (n=9) and spermatozoa (n=5) of infected male partners of the serodiscordant couples was amplified by nested PCR and sequenced. The translated amino acid sequence of these variants shows the presence of distinct HIV variants in PBMCs and sperm of the same individual (Fig. 6). The region 241 to 263 was the predominant conserved region in all the individuals, which corresponds to the C2 region of the env gene. In addition, the small regions also showed the conserved sequence in C2 as well as the V3 region. Moreover, eight out of nine males showed the presence of N-linked glycosylation (NLG) sites in the variants present in their PBMCs while the remaining one male showed the presence of four NLG sites. While four out of five males showed seven NLG sites, the remaining one showed six NLG sites in the HIV variants present in their sperm (Table 3). The presence of NLG sites in the V3 region at position No. 301 suggests the presence of CCR5 tropic variants in PBMCs and sperm samples of these individuals. We have reported the presence of distinct HIV variants following analysis of the translated amino acid sequence of the C2-V3 region of the env gene of HIV-1C in PBMCs and sperm of individuals from concordant couples. 15 However, the variation in sequence of HIV-1C isolates from PBMCs and sperm of the males from concordant couples and discordant couples was found be almost similar, suggesting that the viral variants may not be responsible for prevention of sexual transmission of HIV in these seronegative females.

Translated amino acid sequence of the C2-V3 region of the env gene of HIV-1C from peripheral blood mononuclear cells (PBMCs) and sperm of the HIV-infected males of the serodiscordant couples. The C2 region starts at position No. 218 of the env gene (the amino acid sequence from 218 to 220 not mentioned in this figure). The V3 region is from 296 to 330. The letters underlined represent the constant region while the remaining are the variable regions. The bold letters marked with an asterisk (*) represents NLG sites. PB, PBMCs; SP, spermatozoa.
Participant declined to provide sample.
NLG, N-linked glycosylation; PBMCs, peripheral blood mononuclear cells.
Discussion
HIV is known to bind the conventional CD4 receptor and subsequently transmit the virus into host cells by interaction with CCR5 and/or CXCR4 coreceptors. However, the spermatozoa 18 and vaginal epithelial cells are devoid of the conventional CD4 receptor suggesting the presence of an alternate receptor for HIV binding and entry into these cells. By immunofluorescence we have demonstrated the presence of hMR 10,14 on vaginal epithelial cells, which are the primary sites for male-to-female sexual transmission of HIV. Furthermore, by Western blot, PCR, and real time PCR methods HIV has been shown to bind specifically to hMR on vaginal epithelial cells. 10 HIV binding to hMR has been shown to induce the production of MMP9, which results in the breakdown of the extracellular matrix 10 and therefore entry into the host cells. This study demonstrated immunofluorescent localization of hMR on 90–95% of vaginal epithelial cells of normal adult seronegative females (Fig. 1) from the control group (n=52) and 0–11% of the vaginal epithelial cells of seronegative females from serodiscordant couples (n=39) having unprotected sex with their infected male sexual partner (Fig. 2 and Table 2). Of vaginal epithelial cells of HIV-infected females 8–10% showed the presence of hMR. It has been reported that hMR expressed by vaginal epithelial cells has a high affinity for HIV gp120 and this binding induces production of MMPs, which may lead to degradation of tight junction proteins and the extracellular matrix proteins in the vaginal epithelium and basement membrane. 10 This suggests the possible localization of hMR in a smaller number of vaginal epithelial cells of HIV-infected females.
The nine male sexual partners of the serodiscordant couples who participated in the study were on antiretroviral therapy (ART) for more than 3 years and both the sexual partners were counseled for protected sexual practices with infected partners. However, the safe sex practices were not consistently followed, which is evident from the children born and their age in the case of some of the serodiscordant couples (Table 1). The children born to these couples were seronegative. This suggests the possibility of unprotected sexual practices in some of these couples. Therefore the localization of hMR in a smaller number of vaginal epithelial cells may possibly have prevented HIV binding and entry into these cells and protected these females from sexual transmission of HIV.
The nine serodiscordant couples were further investigated for a possible association of other factors in the prevention of HIV transmission. HIV infects cells by binding of the viral envelope (Env) protein to the host CD4 receptor and then to a coreceptor, most commonly CCR5. The importance of CCR5 in the pathogenesis of HIV-1 is shown by the fact that individuals who are homozygous for an inactivating 32-base pair deletion in CCR5 (CCR5-Δ32) are highly resistant to HIV infection while heterozygotes typically live longer after HIV infection due to reduced levels of CCR5 expression. 19 –24 Limited studies in the Indian population have shown that the rates of CCR5-Δ32 microdeletion have been reported to be very low, in the range of about 0.05% in Rajasthan, 0–0.03% in Andhra-Pradesh, 25 1.5% in North Indians, 26 1–3% in South Indians, 27 and 3% from the control group. 28 The precise association of CCR5-Δ32 microdeletion and prevention of HIV transmission needs to be investigated further in a larger population in India.
In this study the DNA from PBMCs of the nine seronegative females was also tested for deletion of CCR5-Δ32 by PCR amplification of the CCR5 gene using a pair of specific primers flanking for the CCR5-Δ32 mutation. The serodiscordant females investigated did not show the CCR5-Δ32 deletion as evident from a single band of 225 bp (Fig. 5), suggesting the presence of wild-type homozygous CCR5 in these serodiscordant females. This further indicates that these females may possibly be protected due to the expression of hMR in a lower number of vaginal epithelial cells.
Additionally, HIV is known to be genetically extremely variable. Distinct variants have been detected in PBMCs and different tissues such as lymph node, spleen, brain, lung, and urogenital cells and secretions. 29 –34 Our recent studies on the characterization of the C2-V3 region of the env gene of HIV-1C showed the presence of distinct variants in PBMCs and sperm of the same HIV-infected males from concordant couples with variable infectivity and a number of NLG sites, which may influence the viral affinity to host cells and sexual transmission of HIV. This study demonstrated the presence of genotypically distinct HIV variants in PBMCs and sperm of the infected partner of the discordant couples with the presence of the NLG site in the V3 region (Table 3) suggesting the presence of R5 tropic variants in all nine infected male sexual partners. Additionally, their female sexual partners did not show the CCR5-Δ32 deletion and remained seronegative.
The viral variation in sperm and PBMCs of the same seropositive males from discordant as well as from concordant couples reported recently by us 15 was found to be almost similar, further suggesting that viral variation may not be responsible for prevention of sexual transmission of HIV into these females. Interestingly, in one of the males the viral load in blood was undetectable, but the semen samples showed the presence of virus and all nine males are on ART (Table 1). This also suggests that the viral load in blood does not always correlate with that in the semen and ART may not always be effective in the reduction of semen viral load and therefore in the sexual transmission of HIV.
This study demonstrated the presence of hMR in a smaller number of vaginal epithelial cells, which are the predominant site for male-to-female sexual transmission of HIV, and showed that these females remained seronegative despite unprotected sex with their HIV-infected male sexual partner. The precise mechanism of hMR in sexual transmission of HIV is still not completely known and needs to be investigated in a larger population. Associations of hMR in the sexual transmission of HIV also suggest the need for revised strategies for the prevention of sexual transmission of HIV.
Footnotes
Acknowledgments
We are thankful to the NIH AIDS Reference and Reagent Program Bethesda, MD, for supplying reagents. We are grateful to the Indian Council of Medical Research, Government of India, for the Senior Research Fellowship to Shivaji K. Jadhav. We are thankful to Dr. Kenneth H. Mayer and Dr. Deborah J. Anderson for providing Short Term AITRIP Fogarty Training and Fellowship to Shivaji K. Jadhav from Brown/Tufts/Miriam University, Fogarty AITRIP Programme, USA. We also acknowledge the technical support of Ms. Varsha P. Padwal and Ms. Sashaina Fanibunda of the National Institute for Research in Reproductive Health, Mumbai, India. Manuscript no. MS/NIRRH/17/11.
Author Disclosure Statement
No competing financial interests exist.