
Abstract
Drug resistance is a key cause of failed treatment of HIV infection. The efficacy of nonnucleoside reverse transcriptase-inhibiting (NNRTI) drugs is impaired by the rapid emergence of drug-resistant mutations. The literature supports the idea that purposefully designed flexible NNRTIs at an active site may help overcome drug resistance. It is proposed here that the usual “lock and key” model, with respect to NNRTI drug design, be expanded to consider creating “master keys” that would automatically adjust conformations to fit all of the “locks” mutations may make. The present work introduces the novel perspective of designing and creating supramolecular assemblies as potential NNRTIs (instead of the relatively more rigid single-molecule inhibitors). Specifically, flexible self-assembling quinhydrone supramolecular dimers formed from quinonoid monomers (designed to be highly flexible NNRTIs themselves) will be offered as a working example of this new perspective in NNRTI drug design. Quinonoid compounds have demonstrated binding interactions at various sites of the HIV-1 RT enzyme, including the elusive ribonuclease H area. Quinhydrone self-organized dimers have at some point in their molecular architecture a noncovalently interacting donor–acceptor ring pair complex. This complex is at the heart of the increased torsional, rotational, and translational motion this species will experience at a particular active site. Flexible supramolecular assemblies, together with their flexible monomer components, may offer a critical advantage in retaining potency against a wide range of drug-resistant HIV-1 RTs. This new supramolecular perspective may also have broader implications in the general field of antimicrobial drug design.
Introduction
A
A number of pharmacologically active nonnucleoside reverse transcriptase inhibitors (NNRTIs) have been identified. Many of these inhibitors appear highly potent, relatively less toxic, and can specifically inhibit HIV-1 RT. However, the rapid emergence of HIV strains resistant to these compounds in vitro has become a major concern that may affect further development of these types of drugs. 1,6,7 All established drugs that target HIV-1 RT have binding sites either at the polymerase active site, or the nonnucleoside binding pocket, a hydrophobic depression created by the binding of NNRTIs and located near the base of the p66 thumb subdomain, about 10 Å from the polymerase active site. 8 –10 It is therefore not surprising that most of the major known resistance-conferring mutations are also located in the vicinity of the polymerase active site. Drug resistance is a key cause of failure for treatment of HIV infection.
The efficacy of NNRTI drugs is impaired by the rapid emergence of drug-resistance mutations. 1 –7 The literature supports the idea that purposefully designed flexibility of NNRTIs within an active site may help overcome drug resistance. 11,12 Although the general concept associated with the design of flexible RT inhibitors is not new, 11,12 it is important to note that the use of supramolecular complexes in antimicrobial drug design, and in particular anti-HIV drug design, is an important novel extension of this concept. As is described below, self-assembling noncovalently linked nanostructures, in other words, supramolecular nanotechnology, offers a greater advantage in the design and creation of more sophisticated and flexible molecular architectures.
One explanation of the mechanism of drug action (e.g., enzyme inhibitors), known as the receptor site or “lock and key” theory, 13 basically asserts that a drug (the key) combines with a receptor site (lock) to produce a pharmacological effect (e.g., block an enzyme from functioning). Drugs that have the right conformation, and fit into the receptor site, are said to have an “affinity” for that particular receptor site. Only drugs that fit into the receptor site will produce a pharmacological response. Furthermore, through what is often referred to as the “chemical structure–activity relationship,” drug molecules that “fit correctly” have specific chemical structures. The chemical structure of a drug will determine whether a drug molecule (key) will fit and bind in the receptor site (lock) and produce a pharmacological effect, where binding interactions will be dependent on minimizing Gibbs free energies at the site. 13 From this, we may sometimes extrapolate and predict that drugs that are similar in composition and chemical structure may have similar effects. 13
Furthermore, the modification of a drug molecule can influence its pharmacological actions, or alternatively, modification of the active site through mutational resistance of the microorganism may also lead to mutational resistance of a particular drug.
It is proposed here that the usual “lock and key” model of drug design be expanded to consider creating “master keys” that through purposefully designed and controlled adaptability at an active site (flexible NNRTIs), can automatically adjust conformations to fit all of the “locks” mutations may make. The present work introduces the novel perspective of designing and creating supramolecular dimer assemblies as potential NNRTIs (instead of the relatively more rigid single-molecule inhibitors). Specifically, self-assembling quinhydrone supramolecular dimers as NNRTIs, formed from quinonoid monomers designed to be highly flexible NNRTIs themselves, will be offered as a working example of this new perspective in NNRTI drug design. It is the quinhydrone “donor–acceptor complex” that is at the foundation of increased rotational, translational, and torsional degrees of freedom the complex would experience at an active site. Quinonoid (monomer) compounds have demonstrated inhibitory binding on various regions of the HIV-1 RT enzyme, including apparent inhibition in the elusive ribonuclease H (RNase H) area. 14 –21
Two sesquiterpene hydroquinones, peyssonol A and peyssonol B, of the Red Sea algae Peyssonelia species, have been shown to be potent inhibitors of HIV-1 and HIV-2 reverse transcriptase (RTase). 14 These quinonoids behave as noncompetitive RTase inhibitors and repressors of HIV-1 replication. 14 Several quinone, naphthoquinone, and quinonoid-like derivatives have been shown to have varying potency as HIV-1 inhibitors. 15 –18 Quinonoids are well known to take part in redox interactions and have similar redox chemistry and cation metal-chelating properties as other active site RNase H inhibitors. 19 –21 Hydroquinones show limited cytotoxicity and carcinogenicity although bone marrow micronuclei have been reported. 22 Epidemiological studies of hydroquinones have demonstrated lower death rates and reduced cancer incidences among individuals employed in the production of hydroquinones. 22,23
Quinonoid monomers are well known to be able to form unique stable supramolecular dimer assemblies called quinhydrones. 24 –26 Quinhydrone complexes have similar binding affinities as the monomers that form them, but are relatively more stable and can deliver more flexibility and positional adaptability than the constituent monomers at a particular active site. 24 –26 They offer an interesting and important new perspective in NNRTI drug design research with implications for other areas of antimicrobial drug design.
Quinhydrone Supramolecular Complexes
Quinone compounds are known to be able to form supramolecular complexes based on noncovalent interactions. 24 –26 The basic feature of any quinhydrone complex is the stable face-to-face, donor–acceptor, and hydrogen-bonding interactions between a hydroquinone and quinone moiety. The most simple, classic example of a quinhydrone is the stable noncovalently interacting dimer complex formed by p-benzoquinone (cyclohexa-2,5-diene-1,4-dione) with 1,4-hydroquinone (benzene-1,4-diol) (Fig. 1). This compact and stable complex can be synthesized as dark shining needles, with moderate water solubility, from a 1:1 mixing of the reactants. 26 Quinhydrone complexes will form spontaneously through self-assembly interactions on mixing (solution). A myriad of quinhydrones may synthesized by varying functionalities at the carbon centers (Fig. 2). 27 –29
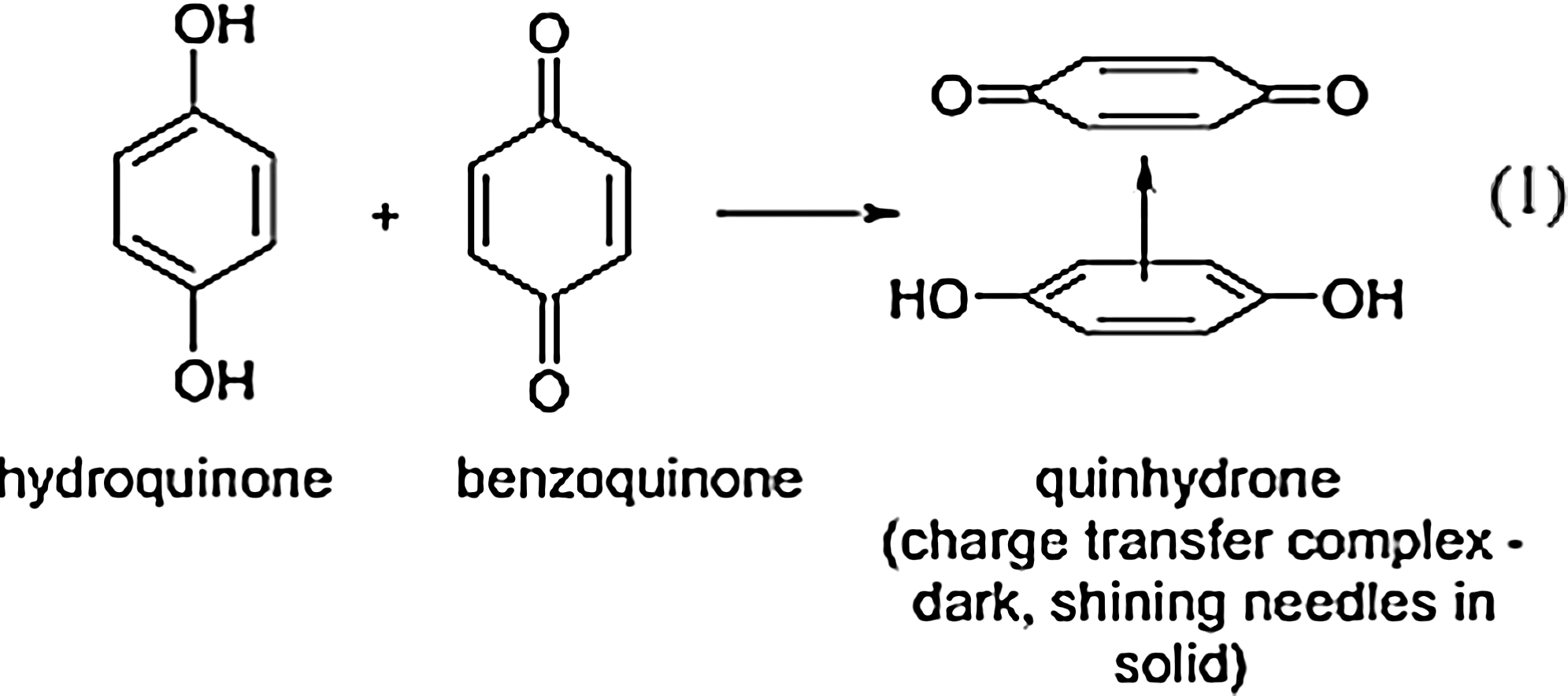
The most basic donor–acceptor supramolecular quinhydrone dimer complex is illustrated. 26
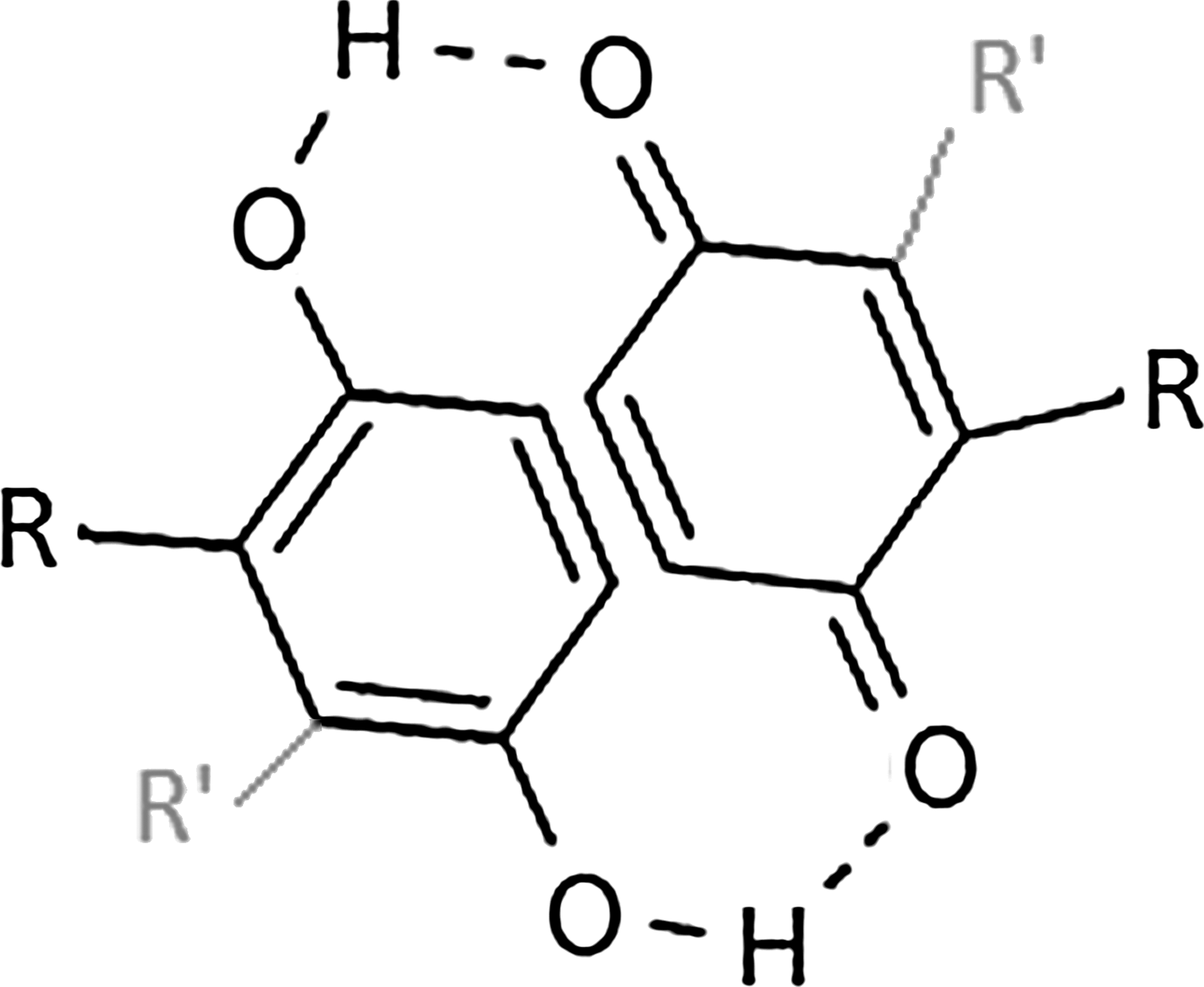
Basic quinhydrone starting structure for the design of novel nonnucleoside reverse transcriptase-inhibitors (NNRTIs). All complexes have a 1:1 ratio of quinone to hydroquine (where R and R′ represent complementary functional groups, which may be varied as desired).
Although some extremely large, stable, and sophisticated quinhydrone supramolecular dimer complexes can be synthesized, the need for such large complex assemblies is not necessary for the purposes of designing quinhydrone NNRTIs. Instead, a design strategy focused on creating flexible, more compact quinhydrone supramolecular structures will be followed. For the purpose of this work, quinhydrone complexes will all contain a 1:1 ratio of quinone to hydroquinone of complementary size and substitution having the following basic structure (Fig. 2). Each of these complexes will have at least one quinhydrone unit (or possibly more than one if desired) in their supramolecular structure. Note that unique functional assemblies may be easily modified by varying functional groups (R and R′ in Fig. 2). Quinhydrone complexes generally have more redox stability, controlled redox reactivity, reliability, and relatively lower toxicity than monomer components. 24 –29 Notably, however, the main characteristic that we exploit here is that the quinhydrone “basic structural unit” or “complex” (Fig. 1) will be the main source of increased torsional, rotational, and translational motion, leading to an NNRTI with increased positional adaptability at an active site. 26
The term “complex” continues to have different connotations in chemistry. Here it has been taken to mean, experimentally, a substance formed by the noncovalent interaction of two component molecules that may reversibly dissociate into components, at least partially under certain conditions. This definition suggests that there is little or no contribution from covalent binding in the ground state. However, it must be recognized that there is a gradient from these weaker interactions that will depend on the strength of π–π electron interactions, hydrogen-bonding interactions, and other factors such as pH, background electrolyte, and solvation factors. These factors will all have to be taken into account when designing new molecular architectures. Quinones are ubiquitous in cellular structures and natural systems; they control photosynthetic electron transfer cycles, important redox interactions in cellular processes, and form the redox active and photoactive portion of humic substances, namely fulvic acid and humic acid, found in all soils and natural waters. 26,30 –35 Quinonoid dimer complexes intended to be used as HIV-1 RT inhibitors will have varied complexity. In this respect, much may be gleaned from the field of supramolecular chemistry.
Supramolecular chemistry is a branch of chemistry concerned with the coalescence of molecules into noncovalent arrays, in other words, molecular recognition. 32,36 Molecular recognition relies on the complementarity of size, shape, and chemical functionalities. It explores and exploits intermolecular forces, the weak attractions that act over short distances between molecules. These forces, hydrogen bonds, aromatic π-stacking, and polar and van der Waals interactions, are the ones that bring molecules together into complexes. In the present case, in order to design quinhydrone complexes for potential use as NNRTIs, one must consider symmetry and complementarity of size and shape in order to form relatively compact, quinonoid complexes that are water soluble, have low toxicity, and will be able to interact at critical active sites of the HIV-1 RT enzyme. The aggregate so formed should have a well-defined structure in solution and be capable of binding behavior that none of its individual components display alone. This definition naturally places an emphasis on three-dimensional assemblies.
Evidence for Conformational Flexibility of Quinhydrone Dimers
Fluorescence line-narrowing (FLN) spectroscopy is an ultralow temperature technique (near absolute zero) in which narrow-banded laser excitation directly into the emitted electronic excited state (S0–S1) is used to reduce the inhomogeneous broadening effects normally encountered in solutions and frozen matrices. 37,38 From a broad distribution of chemically identical but differently oriented and surrounded molecules in the extremely low-temperature solid matrix a subset (“isochromat”) can be selected, for which the S0–S1 (singlet electronic ground–first electronic singlet excited state) energy difference matches the photon energy of the laser (resonance). In all chemical systems tested this usually results in extremely line-narrowed, vibrationally resolved fluorescence spectra, provided that no conformational changes occur during the lifetime of the excited state. 37,38
For the first time, it was reported that a search for fluorescence line-narrowing to obtain vibrational information from near 10 K laser experiments was not successful. 26 Because these extremely low-temperature experiments were not successful in obtaining line-narrowed spectra, it was concluded that enough conformational change was still occurring at 10 K that broadening effects were still occurring. 26 The systems being studied were those of simple quinhydrone complexes in solution (discussed later; and see Figs. 1 and 4). It was determined from this work that the noncovalently interacting donor–acceptor quinhydrone complex was responsible for generating relatively higher reorganizational degrees of freedom at 10 K. These quinhydrone solutions were prepared at various pH levels and background electrolyte concentrations (some similar to biological systems) and remained stable for up to 10 days. 26
It is not difficult to imagine that such complexes may prove to be a useful and interesting starting point to test the hypothesis that more flexible NNRTIs may be more effective at overcoming some of the mutational resistance of HIV. Enhanced conformational freedom, within the boundaries of an active site, may increase the probability that NNRTIs will retain potency relative to more rigid NNRTIs. 11,12
Fortunately, there is no need to perform such extremely low-temperature experiments in order to prove a supramolecular complex has formed. Methods in supramolecular chemistry are sufficiently advanced that a plethora of experimental methods are available to characterize any given supramolecular system. 36 Supramolecular assembly complexes often form new electronic absorption bands, and some of these new bands may be broad, featureless, and reach far into the red, as would be expected for complex assemblies with increased conformational freedom. Indeed, the small, simple quinhydrone supramolecular polymers described above are able to mimic the fluorescence emission spectra and portions of the ultraviolet–visible spectra of the heterogeneous humic polymer mixture known as fulvic acid. 26 This is remarkable because fulvic acid is a complex heterogeneous mixture of polyelectrolyte macromolecules with varying organic functional groups (phenols, quinones, carboxylic acids, aldehydes, carbohydrates, and ketones), and has a molecular weight range from a few hundred to a several thousand grams per mole. 26,33 –35 Supramolecular complexes are often able to mimic the spectroscopic behavior of much larger and complex systems. 36
Design Strategy
As an example, a good starting point would be the structure of a known quinonoid HIV inhibitor, 16 from which a preliminary working model quinonoid may be built (Fig. 3). Figure 3 is representative of one of the smaller molecular weight hydroquinone polymers (so-called humic-like polymers) described in the literature as having anti-HIV activity. 16 This monomer construct was built by substituting R (Fig. 2) with alternating hydrogen atom or hydroxyl (OH) groups and R′ (Fig. 2) with another hydroquinone. The hydroquinonoid monomer may then self-associate into a novel quinhydrone dimer complex (Fig. 4) if the necessary complementary quinonoid monomer is made available to it.
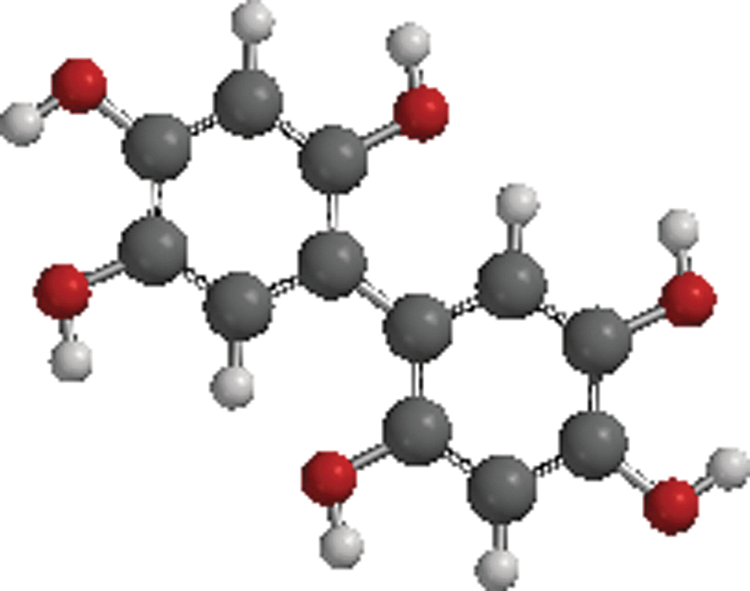
Representative model for a novel quinhydrone monomer [quantum chemical PM3 semiempirical geometric optimization calculations performed with Spartan′10 for Linux/Windows (Wavefunction, Irvine, CA)]. Here the aromatic rings are illustrated as black spheres (carbon atoms), red spheres (oxygen atoms), and white spheres (hydrogen atoms).
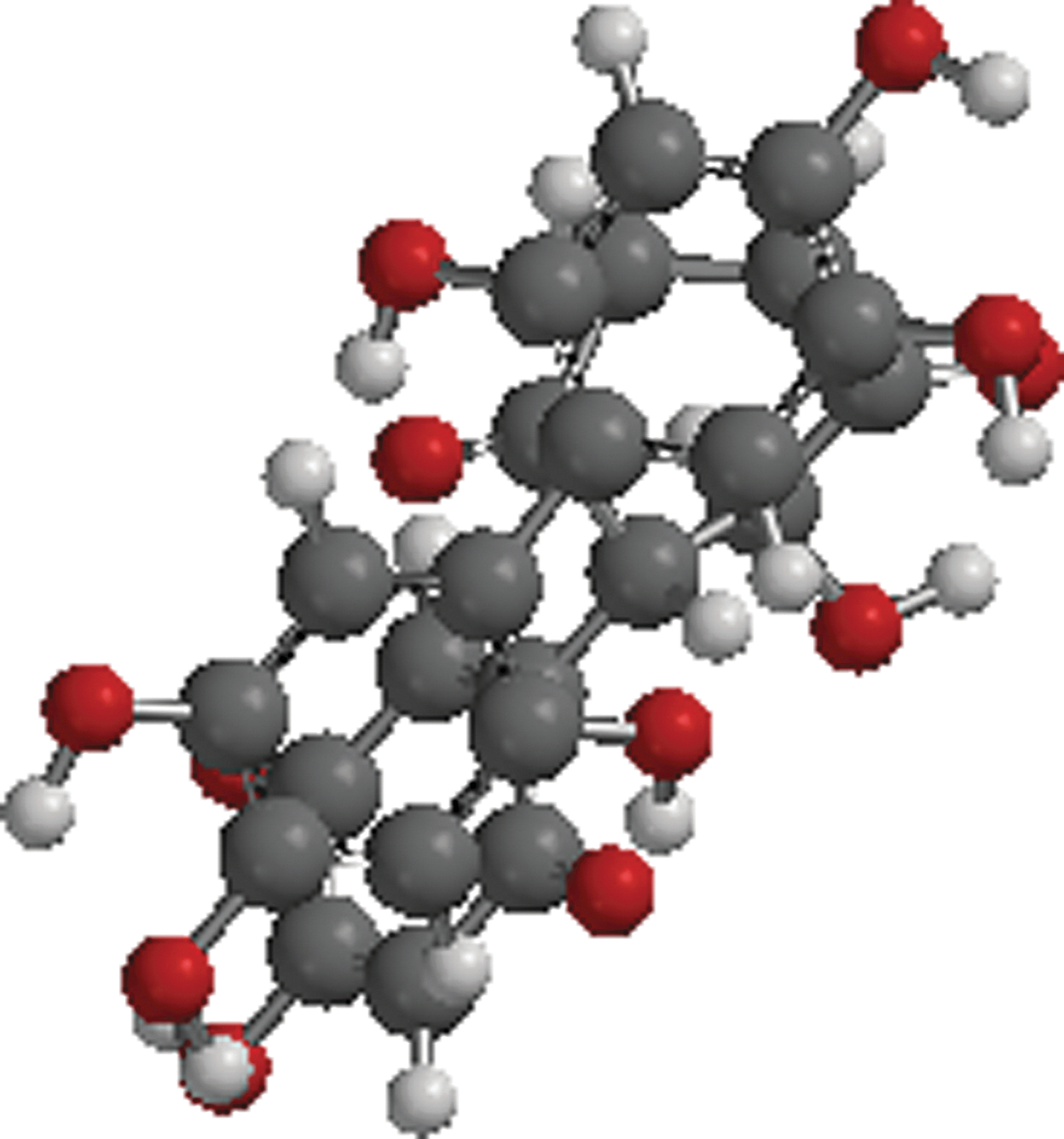
Representative self-assembled quinhydrone dimer pseudo-sphere complex formed by complementary donor–acceptor monomer (Fig. 3) interactions [quantum chemical PM3 semiempirical geometric optimization calculations performed with Spartan′10 for Linux/Windows (Wavefunction)]. Black spheres represent carbon atoms; red spheres represent oxygen atoms; and hydrogen atoms are represented by white spheres.
Complementarity of size, shape, and chemical functionality strongly influences molecular assemblies. Consideration must be made in terms of placing functional groups capable of strategic hydrogen-bonding interactions, as well as aromatic ring effects and other nonbonding interactions. Assuming the three-dimensional shape/size and chemical environment of the active site to be targeted are known, working models may be constructed to fit into specific HIV-1 RT active sites. For example, designing monomers of complementary size and shape with alternate regions of hot (slightly higher) and cold (slightly lower) electron density increases self-association interactions, leading to a more stable dimer complex through complementary electrostatic potential interactions (Fig. 5).
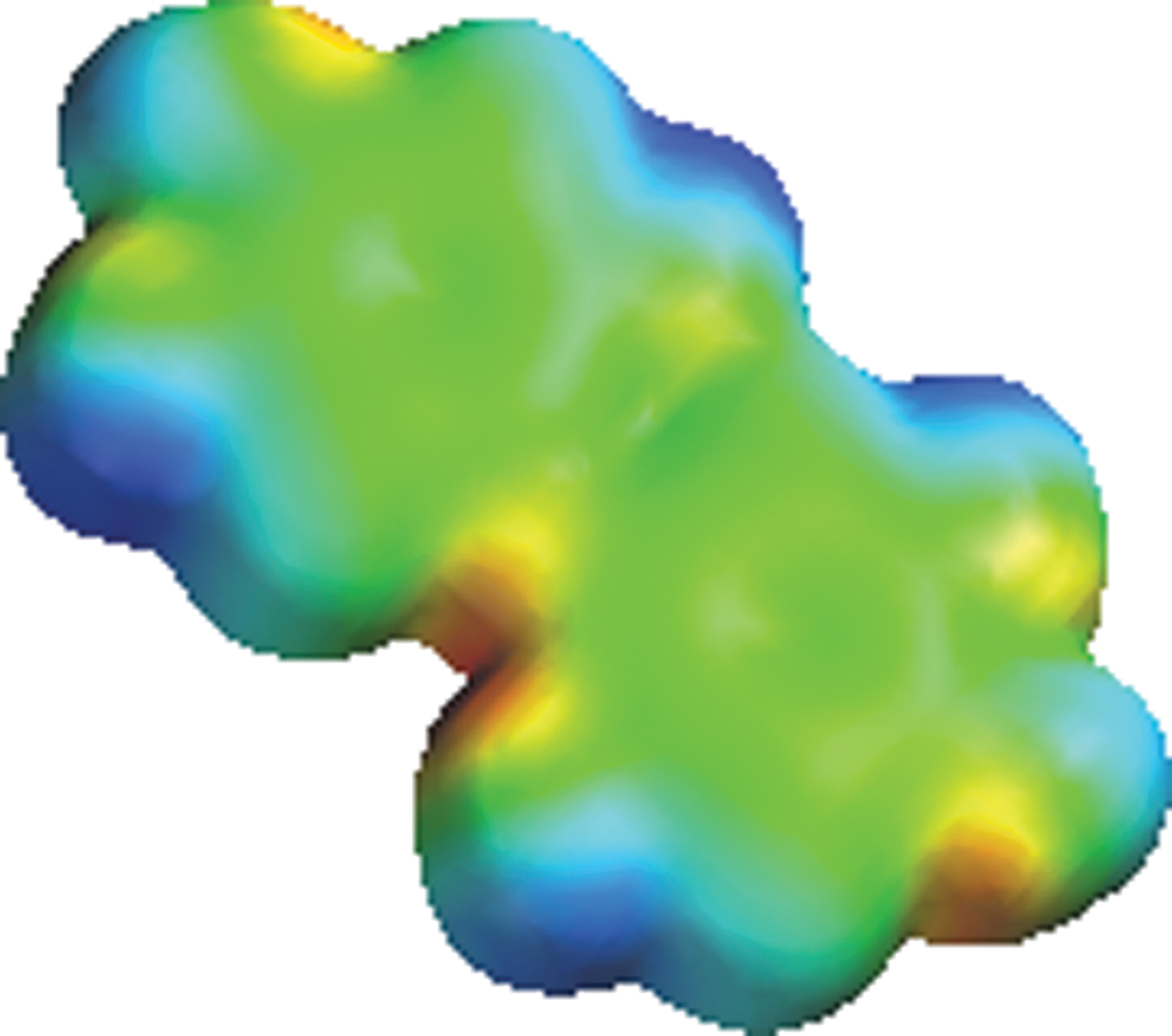
The electrostatic potential has been mapped onto the electron density surface; showing “hot” (red) and “cold” (blue) regions “picketed” along the edge of the half–tennis ball quinonoid monomer structure (two-ring monomer structure from Fig. 3). Creating alternating “hot” and “cold” regions along the edge of the molecule increases self-association potential. This figure was generated on the basis of a semiempirical PM3 quantum chemical calculation and with calculated electrostatic potential mapped onto the electron density surface [quantum chemical PM3 semiempirical geometric optimization calculations performed with Spartan′10 for Linux/Windows (Wavefunction)].
This molecular design is intended to set up noncovalent interactions that complement and strengthen the donor–acceptor quinhydrone interaction. This can be accomplished through a careful choice of added chemical functionalities. In the present case, much use has been made of OH functionalities. Their strategic placement at free carbon centers creates increased hydrogen bonding, increased water solubility, and sets up potential noncovalent interactions with critical amino acid residues at an active site.
As mentioned previously, flexibility of the monomers will also be an important factor to consider, 11,12 as well as designing quinhydrone monomers to be able to function as NNRTIs themselves. For example, larger more flexible monomers can be designed by substituting R (Fig. 2) with two covalently linked hydroquinones having carbon or oxygen atom spacers between the aromatic rings. This would lead to a monomer with three hydroquinone rings attached by carbon or oxygen atoms, allowing for free rotation of the rings. Note the “horseshoe” conformation (Fig. 6A) after geometric optimization calculations have been performed; this shape is a recurring theme with many NNRTIs. 11

Quinhydrones often form pseudo-sphere assembly structures (Fig. 6B) that can undergo binding interactions; however, if divalent cation interactions are desired near the RNase site, for example, then some consideration must also be made of the electrostatic reduction potential of the quinhydrone complex. In this case, adjustments can be made by fine-tuning functional group attachments around the periphery of the complex. Quantum chemical electrostatic potential calculations correlated to HOMO/LUMO (highest occupied molecular orbital/lowest unoccupied molecular orbital) calculations and cross-correlated to experimental cyclic voltammetry and electrochemistry experiments can give the molecular designer important clues in order to optimize the molecular architecture for a desired quinhydrone reduction potential. 24,25,36 Thus designing quinhydrone/quinonoid species as NNRTIs must involve three-dimensional and electrostatic potential considerations.
When designing and studying preliminary quinhydrone structures, it is important to note that all of these molecular moieties undergo strong noncovalent and electron donor interactions. In this case, therefore, it is important that geometric optimization molecular modeling calculations be performed at the semiempirical quantum mechanical level (not molecular mechanics). This is because quantum mechanical (QM) methods treat electrons explicitly and such methods are much better suited for chemical species that undergo complex donor–acceptor and noncovalent interactions. 33 However, QM/MM (quantum mechanical and molecular mechanics) mixed methods may be used to model binding interactions between quinhydrone complexes and the HIV-1 RT enzyme. When dealing with chemical species that are capable of electron donor–acceptor complexes and/or strong noncovalent interactions it is best to use QM/MM mixed methods simultaneously in order to exploit the benefits of both the QM and MM methods. 33 Here the region of most interest in the system (i.e., where the NNRTI binding/displacement/bond breaking/forming is taking place in the enzyme, along with selected surrounding catalytically relevant residues) is best treated with a QM method (e.g., semiempirical calculations). The rest of the enzyme may be treated with a MM force field.
As was mentioned previously, a key feature of the molecular architecture of quinhydrone self-organized dimers is the noncovalently interacting donor–acceptor complex. Quinhydrone complexes may be designed to be stable, but conceivably the monomers that form the dimer may be designed to act as individual HIV inhibitors themselves, as a percentage may dissociate in solution. The key here will be to design a flexible monomer and dimer that will have good positional adaptability within an active site, thus improving resistance to mutational conformational changes at an active site. The work of Das and colleagues 11 and Ohtaka and colleagues 12 supports this hypothesis. In a review presented by Das and colleagues 11 the potency of relatively rigid inhibitors such as nevirapine and α-anilinophenylacetamide (α-APA), which interact extensively with the aromatic side chains of Y181 and Y188, is severely impaired by these two mutations. Das and colleagues 11 reviewed the measured potencies of the NNRTIs against recombinant clinical isolates, and correlated structural and molecular modeling data. Das and colleagues 11 concluded that the conformational flexibility of an NNRTI was important with regard to its ability to retain its potency as an NNRTI through mutational resistance of HIV strains.
The classic lock-and-key model used in inhibitor drug design requires that a potential inhibitor assume a three-dimensional conformation that is complementary to the intended binding site's conformational shape and chemical functionalities. A successful NNRTI should be able to adapt to the mutational related conformational changes within a binding site through conformational repositioning (all within the limits of the lock-and-key model). Figure 7 illustrates the increased positional adaptability a quinhydrone structure may have in comparison with a single-molecule (more rigid) NNRTI in an active site. By virtue of the quinhydrone functionality, a quinhydrone-based NNRTI may be able to reposition and reorient itself more easily around a mutation-based conformational change in an active site. The quinhydrone functionality will have relatively more torsional, rotational, and translational (axial and equatorial) degrees of freedom, effectively acting as a type of compressed molecular “spring” or “shock absorber” within the site.

A rigid inhibitor (left) may be less capable of repositioning itself and may not bind as favorably if a mutation occurs (red star) in an active site for which it was designed. The quinhydrone functionality center (right) may have an advantage through its relatively higher flexibility (increased degrees of freedom for torsional, rotational, and translational motion).
NNRTI Interactions and Inhibition of RT Activity
Various HIV inhibitor studies have shown that NNRTIs are chemically diverse, 1 –8 where one class of NNRTIs may be considerably different from another class in terms of its chemical composition and size. Structural studies together with biochemical work have shown that several NNRTIs bind to the so-called “hydrophobic pocket” located in the p66 subunit of HIV-1 RT at approximately 10 Å from the DNA polymerase active site. 39 This pocket is not observed in the inhibitor-free structure of RT. It is created in the p66 subunit only after the inhibitor has been complexed with the RT. As this pocket is absent in the p51 subunit, these inhibitors cannot bind to the smaller HIV-1 RT subunit. The NNRTI-binding site could best be described as a flexible domain rich in hydrophobic amino acids that would open only in the presence of the inhibitor. Although a major portion of the pocket is predominated by hydrophobic amino acid side chains, at the entrance of the pocket there are hydrophilic residues Lys-101 and Lys-103 from p66 and Glu-138 from the p51 subunit. Some of the amino acids involved in the pocket (or in close vicinity to it), are Tyr-181 and Tyr-188, Val-106, Phe-227, Trp-229, Leu-100, Val-179, Gly-190, Leu-234, His-235, Pro-236, and Tyr-318. In particular, Tyr-181 and Tyr-188 have been found to be critical for the binding of many NNRTIs. The absence of tyrosine residues 181 and 188 in HIV-2 RT explains the lack of effect of NNRTIs on this RT. 39 Although Tyr-181 and Tyr-188 lie within a conserved motif, they are not essential for the polymerase activity. Hence, when a nonnucleoside compound challenges the enzyme, HIV-1 RT mutations at the Tyr-181 and Tyr-188 residues emerge that allow the enzyme to readily circumvent the inhibitory action of the nonnucleoside agents. All of these factors must be taken into account when designing quinhydrone-based NNRTIs.
Notably, NNRTIs have been found to bind to RT in different modes 40 –43 and the flexibility of the HIV-1 reverse transcriptase hydrophobic pocket accommodates NNRTIs of different shapes and sizes. In spite of different chemical structures, many assume a “butterfly-like” shape 40 –43 or so-called “horseshoe” conformation. 11 Interestingly, these conformations are similar to those formed by quinonoid polymer complexes (monomer; Fig. 6A). 16 These monomers (Fig. 6A) may self-associate to form dimer complexes as illustrated in Fig. 6B. The hydrophobic pocket, as well as other areas of the HIV-1 RT enzyme, could accommodate such quinhydrone structures (Fig. 8A–C).

As was mentioned earlier, several quinonoids have been reported to behave as NNRTIs. 14 –21 with some appearing to favor RNase binding. 17 As quinonoids and quinhydrones are well known to specialize in similar redox interactions with divalent metal cations, 24 –31 it is conceivable that increasing efficiency of binding interactions at the RNase H active site may involve two simultaneous binding mechanisms: (1) metal ion binding interactions and (2) amino acid residue interactions. For example, using a known quinonoid NNRTI as a starting point and modeling its interaction on the HIV-1 RT surface, we observe some interesting preferred predicted interactions. A quinhydrone (Fig. 4) formed from a known quinonoid NNRTI starting structure 16 (Fig. 3) illustrates that metal ion Mg2+ coordination displacement takes place near the catalytically essential acidic residue Asp-443 and aromatic residues Tyr-457 and Tyr-441 in the RNase H active site (Fig. 8A and B). Similar quinonoid polymer moieties in coordination with the metal ion cofactor (Mg2+) and aromatic π–π stacking interactions with the important conserved aromatic amino acid residue Tyr-501 were also observed (data not illustrated).
Furthermore, considering that the topography of the RNase H domain is a relatively flat surface, 20 unlike the polymerase domain, which contains mobile subdomains and hydrophobic pockets that provide a foothold for small molecules, a slightly larger quinhydrone (Fig. 6B) was predicted to preferentially interact in this region. Molecular modeling calculations also suggest binding interactions may indeed be predicted near the Trp-229, Met-230, Gly-231, Tyr-232 quartet (Fig. 8C), which is important for the DNA polymerase and RNase H domains.
Notably, Trp-229 would be an ideal target site, as an important characteristic of the hydrophobic pocket is that Trp-229, Phe-227, and Leu-234 are conserved in HIV-1 RT and HIV-2 RT enzymes 44 and in vitro mutation of Trp-229 in HIV-1 RT has been shown to significantly impair DNA polymerase activity. 45 Similar calculations, using the quinhydrone structure from Fig. 4, hint that quinhydrone-binding interactions may also be predicted to occur near Trp-266 in the minor groove-binding track. Such displacement of critical metal ions and conformational changes in the HIV-1 RT enzyme may explain some of the apparent inhibitory capacity of quinonoid chemical species, as well as possibly predict the potential of quinhydrone derivatives as NNRTIs.
Although the RT of HIV has been a main target for antiviral drugs, almost all compounds developed so far inhibit the polymerase function of the enzyme. Few antiviral agents inhibit specifically the necessary RNase H function of RT. 20 This extreme bias of drugs toward the polymerase activity could be attributed, in part, to the relatively flat surface topography of the RNase H domain. 20 Taking advantage of the divalent cation metal-binding interactions of quinhydrones/quinonoids and targeting simultaneous cooperative binding with critical amino acid residues may lead to novel potent RNase inhibitors. Interestingly, molecular modeling of quinhydrones (Figs. 4 and 6B) suggests that some sort of twisting and distortion of the quinhydrone complex occurs, along with simultaneous complex binding interactions only at certain preferred sites of the HIV-1 RT enzyme (near critical cation metal ions and specific amino acid residues; Fig. 8A–C). Twisting and distortion of quinhydrone complexes are often associated with electron charge transfer type interactions between the complex and a third species. 25,33
Flexibility of NNRTIs appears to be important. It has been proposed that some inhibitors, for example, the DAPY (diarylpyrimidine) inhibitors TMC120 and TMC125, 43 efficiently inhibit the L100I mutant HIV-1 RT because they have torsional flexibility, 11 which allows them to bind effectively to the different conformations of the hydrophobic pocket found in the wild-type HIV-1 RT and the drug-resistant mutants. Quinhydrone/quinonoid complexes would appear to be capable of similar conformational adaptation.
Interestingly, preliminary results indicate that quinonoid polymers similar to those described previously, 26 and illustrated in Figs. 4 and 6, appear to reduce both HIV-1 and FIV reverse transcriptase (RTase) activity in a concentration-dependent manner in infected cultures, while also suppressing HIV-induced syncytium formation. 46 Moreover, HIV-1 pseudotyped virus infection was also apparently reduced by these quinonoid polymers, together with a reduction in p24 expression and direct inhibition of RTase activity. 46 Treatment of human cells with the polymers demonstrated drug accumulation in cystolic compartments with limited cytotoxicity at supratherapeutic concentrations. 46 Moreover, polymers similar to those illustrated in Fig. 4 (MT-2 cell, RT activity assay) appeared to inhibit nucleoside-resistant virus families K65R, K103N, and nonnucleoside-resistant virus family D30N (mutations associated with drug resistance).
Conclusions
Emergence of drug resistance in patients treated with reverse transcriptase inhibitors is a major limitation of antiviral therapy. 1,20 The thermodynamic study performed by Ohtaka and colleagues 12 predicted that flexibility of a protease inhibitor was essential for its favorable binding to mutant enzymes. Later, Das and colleagues 11 conducted crystal structure and molecular modeling analyses and suggested a role for conformational flexibility in compensating for the effects of NNRTI resistance mutations. Molecules of high torsional flexibility and positional adaptability (translation and rotation) that are capable of undergoing complex noncovalent binding interactions may be able to overcome some of the mutational resistance of HIV-1 strains. 11,12 Design features of an effective inhibitor against a mutating target should include such factors as the physical and chemical changes of the pocket caused by mutations, conformational-elastic limits of the pocket, type and extent of interactions of an inhibitor with different parts of the pocket, locations of conserved and mutated amino acid residues in the pocket, mode of inhibition, and the role of resistance mutations. Although the inhibitor should be flexible so that it can bind in the modified pockets of a mutant target, important inhibitor–target interactions should not be adversely affected by inhibitor flexibility. The use of supramolecular complexes, and in particular supramolecular quinhydrone dimers, may offer a useful new perspective in NNRTI drug design and may even have broader implications in the field of antimicrobial drug design.
Notably, although the present work describes the novel concept of using supramolecular nanostructure quinonoids as antimicrobials (particularly HIV inhibitors), there are a plethora of alternative molecular nanostructures that may be considered in the design of NNRTIs. Supramolecular chemistry in antimicrobial drug design should not be limited to quinonoid structures alone.
In a sense, supramolecular chemistry is being used to design and create tiny self-assembled molecular machines (NNRTIs) to disable larger self-assembled molecular machines (HIV strains).
Footnotes
Acknowledgments
The author thanks Canadian University College for support, and Dr. Mark Wainberg (McGill University) for many fruitful conversations and laboratory testing (McGill AIDS Centre).
Author Disclosure Statement
No competing financial interests exist.