
Abstract
Development of an effective low-cost anti-acquired immunodeficiency syndrome (AIDS) drugs is needed for treatment of AIDS patients in developing countries. Host cell lipid raft microdomains, which are enriched with cholesterol, glycolipids, ceramide, and gangliosides, are important for human immunodeficiency virus type 1 (HIV-1) entry. Retinoid analogs have been shown to modulate ceramide levels in the cell membrane, while cholera toxin B subunit (CT-B) specifically binds to the ganglioside GM1. In this study, we found that the acyclic retinoid analogs geranylgeranoic acid (GGA) and NIK-333 as well as CT-B efficiently attenuate CXCR4-tropic, but not CCR5-tropic, HIV-1 vector infection. We also found that GGA and NIK-333 suppress CXCR4-tropic HIV-1 infection by attenuating CXCR4 expression. CT-B also attenuated CXCR4-tropic HIV-1 infection, but did not suppress CXCR4 expression. These results suggest a distinct role for lipid raft microdomains in CXCR4- and CCR5-tropic HIV-1 infections and illuminate novel agents for the development of AIDS therapy.
Introduction
H
Lipid raft microdomains of target cell membranes are required for HIV-1 infection. 2 –6 Lipid rafts are enriched with cholesterol, glycolipids, and ceramide. 7 Extraction of cholesterol from cell membranes, 4,6 binding of cholesterol with various factors, 2,8 and inhibition of biosynthesis of cholesterol 9,10 or glycolipids 11 –13 suppress HIV-1 infection, suggesting that cholesterol and glycolipids may be targets for novel anti-HIV-1 drugs. In this study, we examined the effects of lipid raft-associated factors, which were isolated from natural products, on HIV-1 vector infection.
Retinoic acid and its analogs modulate ceramide levels in cell membranes. 14 –20 Retinoid analogs may inhibit HIV-1 infection by altering ceramide levels of the target cell membrane. In fact, an all-trans retinoic acid 21 and a weak nuclear retinoid receptor agonist, N-(4-hydroxyphenyl) retinamide (4-HPR), 22 inhibit HIV-1 infection 19, 23 ; however, because 4-HPR has severe toxicities, such as induction of vitamin A deficiency symptoms, clinical application of 4-HPR is restricted. 24 Geranylgeranoic acid (GGA), which is a natural acyclic retinoid analog present in medicinal herbs, 25 serves as a weak agonist for retinoid receptors, similar to 4-HPR. 26, 27 NIK-333, which is an artificial acyclic retinoid analog with a structure similar to GGA (Fig. 1), prevents recurrence of hepatocellular carcinoma following oral administration without any obvious side effects in clinical studies of liver cancer patients. 28,29 We analyzed the effects of the acyclic retinoid analogs GGA and NIK-333 on HIV-1 vector infection.
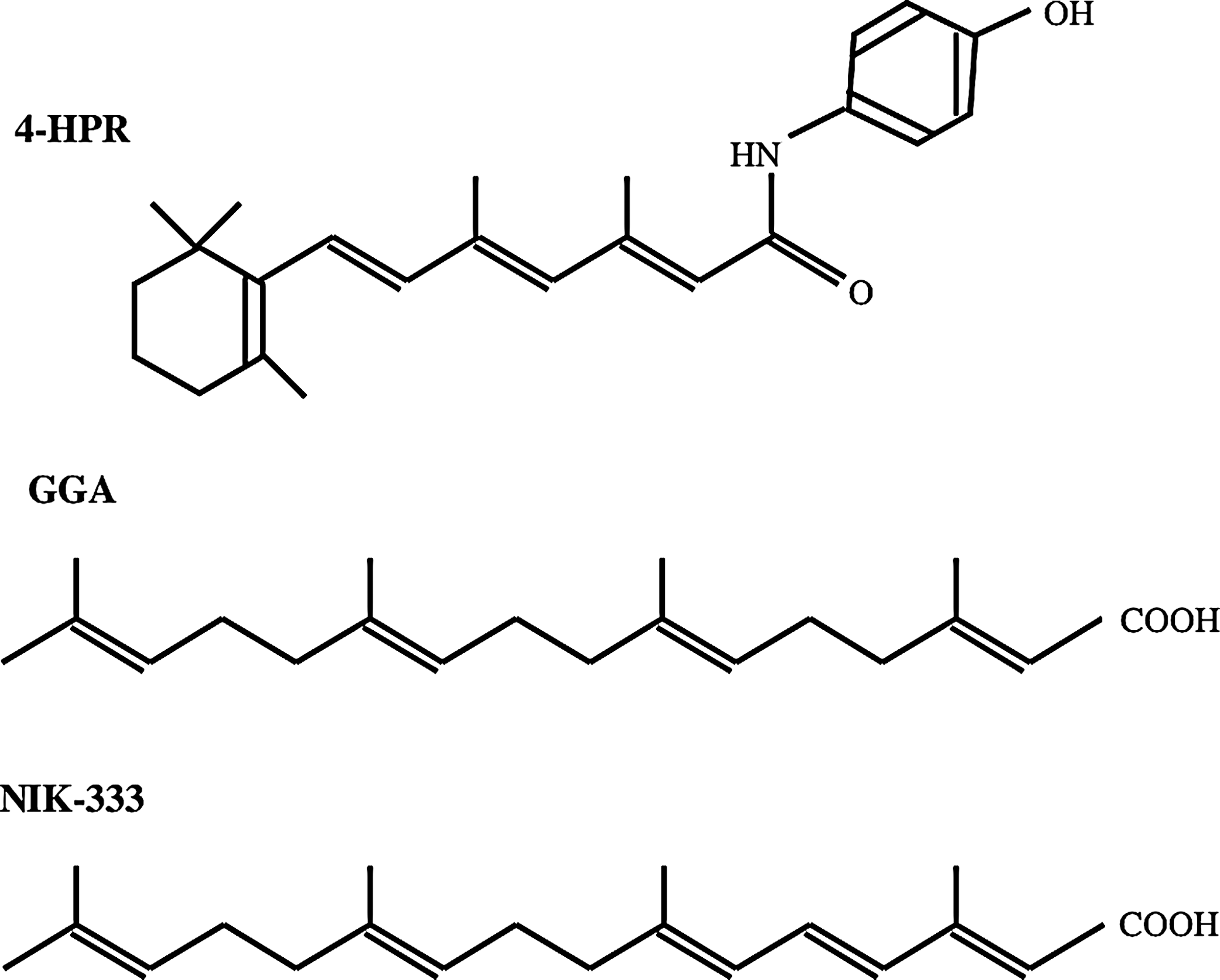
Chemical structures of 4-HPR, GGA, and NIK-333.
Cholesterol is enriched in lipid raft microdomains and requires their structural maintenance. Extraction of cholesterol from cell membranes by methyl-β-cyclodextrin (MβCD), 4, 6 inhibition of cholesterol synthesis by statin, 9,10 or binding of amphotericin B methyl ester to cholesterol 8 suppresses HIV-1 infection. Plant sterols are cholesterol analogs that reduce serum cholesterol levels by replacing cholesterol. 30 Therefore, plant sterols may function as anti-HIV-1 agents.
Because cholera toxin B subunit (CT-B) specifically binds to the ganglioside GM1, this subunit is frequently used as a lipid raft marker. 4,6 The cytopathic determinant of cholera toxin is subunit A, which has the poly(ADP) ribosylation activity of G-proteins. 31 In contrast, the B subunit has no cytopathic effect. GM1 is enriched in raft microdomains and has been reported to bind HIV-1 envelope (Env) glycoprotein. 32 Additionally, CD4-positive lymphocytes that have elevated levels of another gangliosides, GM3, are highly susceptible to HIV-1 fusion and entry. 11 Therefore, CT-B may inhibit HIV-1 infection without cytopathic effects.
In this study, we examined the effects of these raft-associated factors on HIV-1 vector infection. Our results showed that acyclic retinoid analogs and CT-B efficiently suppressed CXCR4-tropic HIV-1 vector infection, providing novel strategies for the development of CT-B or acyclic retinoid analog treatment for AIDS patients. In contrast, these factors did not affect CCR5-tropic HIV-1 vector infection, suggesting that raft microdomains are involved differently in CXCR4- and CCR5-tropic HIV-1 infections.
Materials and Methods
Cells
COS7, 293T, NP2, TE671, and HeLa cells were cultured in Dulbecco's modified Eagle's medium (D-MEM) (Wako) supplemented with 8% fetal bovine serum (Biosource) at 37°C in 5% CO2. NP2 cells expressing CD4 and CXCR4 (NP2/CD4/X4) or CD4 and CCR5 (NP2/CD4/R5) were kindly provided by Dr. H. Hoshino. 33 NP2 cells expressing CD4 and C-terminally HA-tagged CXCR4 (NP2/CD4/X4-HA) were constructed as previously reported. 34 TE671, HeLa, and 293T cells expressing CD4 (TE671/CD4, HeLa/CD4, and 293T/CD4) were constructed with a CD4-encoding murine leukemia virus (MLV) vector as previously reported. 35 MAGIC5 cells, which are derived from HeLa cells, express CD4 and CCR5 and contain the β-galactosidase (β-Gal) gene under control of the HIV-1 long terminal repeat. 36
Expression plasmids
CXCR4-tropic HXB2 and CCR5-tropic JRFL HIV-1 Env expression plasmids were kindly provided by Dr. Y. Yokomaku (National Hospital Organization Nagoya Medical Center). A VSV-G expression plasmid and expression plasmids required for LacZ reporter gene-containing HIV-1 vector construction were obtained from Invitrogen. An expression plasmid encoding C-terminally HA-tagged CXCR4 was constructed as already reported. 34
Transduction assay
To obtain HIV-1 vector particles, COS7 cells were transfected with the HIV-1 vector construction plasmids using Fugene transfection reagent (Roche). The transfected cells were washed with D-MEM medium 24 h after transfection and maintained in fresh medium for 24 h. Target cells were either left untreated or pretreated with the retinoid analogs 4-HPR (Sigma-Aldrich), GGA, or NIK-333 for 2 days or with CT-B (Sigma-Aldrich) or stigmasterol (Sigma-Aldrich) for 1 day. GGA and NIK-333 were synthesized by Kowa Company, Ltd. (Tokyo, Japan). The cells were inoculated with culture supernatants from the transfected COS7 cells and then stained with 5-bromo-4-chloro-3-indolyl-β-
Flow cytometry
To analyze cell surface CD4 expression, suspended cells were either left untreated or treated with an anti-CD4 antibody conjugated with FITC (Sigma-Aldrich). Cell surface expression of CXCR4 or CCR5 was analyzed in suspended cells treated with rat anti-CXCR4 (A80) or anti-CCR5 (T312) monoclonal antibody. 37 As a control, cells were treated with a rat serum. The cells were then washed three times with phosphate-buffered saline (PBS) and treated with an FITC-conjugated anti-rat IgG antibody (Sigma-Aldrich). The stained cells were quantified using a flow cytometer (BD Biosciences).
Western immunoblotting
NP2/CD4/X4-HA cells were treated with the retinoid analogs, and cell lysates were prepared. The cell lysates were subjected to SDS polyacrylamide gel electrophoresis (Bio-Rad) and transferred onto a PVDF membrane (Millipore). The membrane was treated with a mouse anti-HA monoclonal antibody (Covance), and then with an HRP-conjugated anti-mouse IgG antibody (Bio-Rad).
Vector particle binding to target cells
Target cells were incubated with culture supernatants from the HIV-1 vector-producing cells for 1 h at 4°C. The cells were washed three times with PBS, and cell lysates were prepared. HIV-1 Gag p24 levels were measured with a p24 enzyme-linked immunosorbent assay (ELISA) (ZeptoMetrix) to estimate the numbers of HIV-1 vector particles bound to the target cells.
Cell fusion assay
The 293T cells were transfected with the HXB2 Env expression plasmid, which also encodes the Tat protein. As a control, 293T cells were transfected with a Tat expression plasmid. The transfected cells were cultured with MAGIC5 cells 24 h after transfection, and cell lysates were prepared from the cells 24 h after the mixed culture. Upon cell fusion, the Tat protein induced β-Gal expression. β-Gal activity in the cell lysates was measured to estimate cell fusion capability.
Statistical analysis
Differences between two groups were determined by the Student's t-test. The difference was considered statistically significant if the p-value was <0.05 for all tests.
Results
Acyclic retinoid analogs and CT-B inhibit CXCR4-tropic HIV-1 vector infection
To assess whether retinoid analogs inhibit HIV-1 vector infection, target cells were pretreated with 4-HPR, GGA, or NIK-333 for 2 days. The chemical structures of the analogs are shown in Fig. 1. NP2 cells expressing CD4 and CXCR4 (NP2/CD4/X4), NP2 cells expressing CD4 and CCR5 (NP2/CD4/R5), 33 and HeLa cells expressing CD4 (HeLa/CD4) 35 were used as target cells. All of the retinoid analogs inhibited infection by a CXCR4-tropic HXB2 Env-carrying HIV-1 vector (Fig. 2A). Previous reports indicated that 4-HPR inhibits HIV-1 infection, 23 and this result is consistent with our findings. In addition, cell viability was not affected by the analog treatment under these conditions. These results indicate that the acyclic retinoid analogs GGA and NIK-333 as well as 4-HPR inhibit CXCR4-tropic HIV-1 infection.
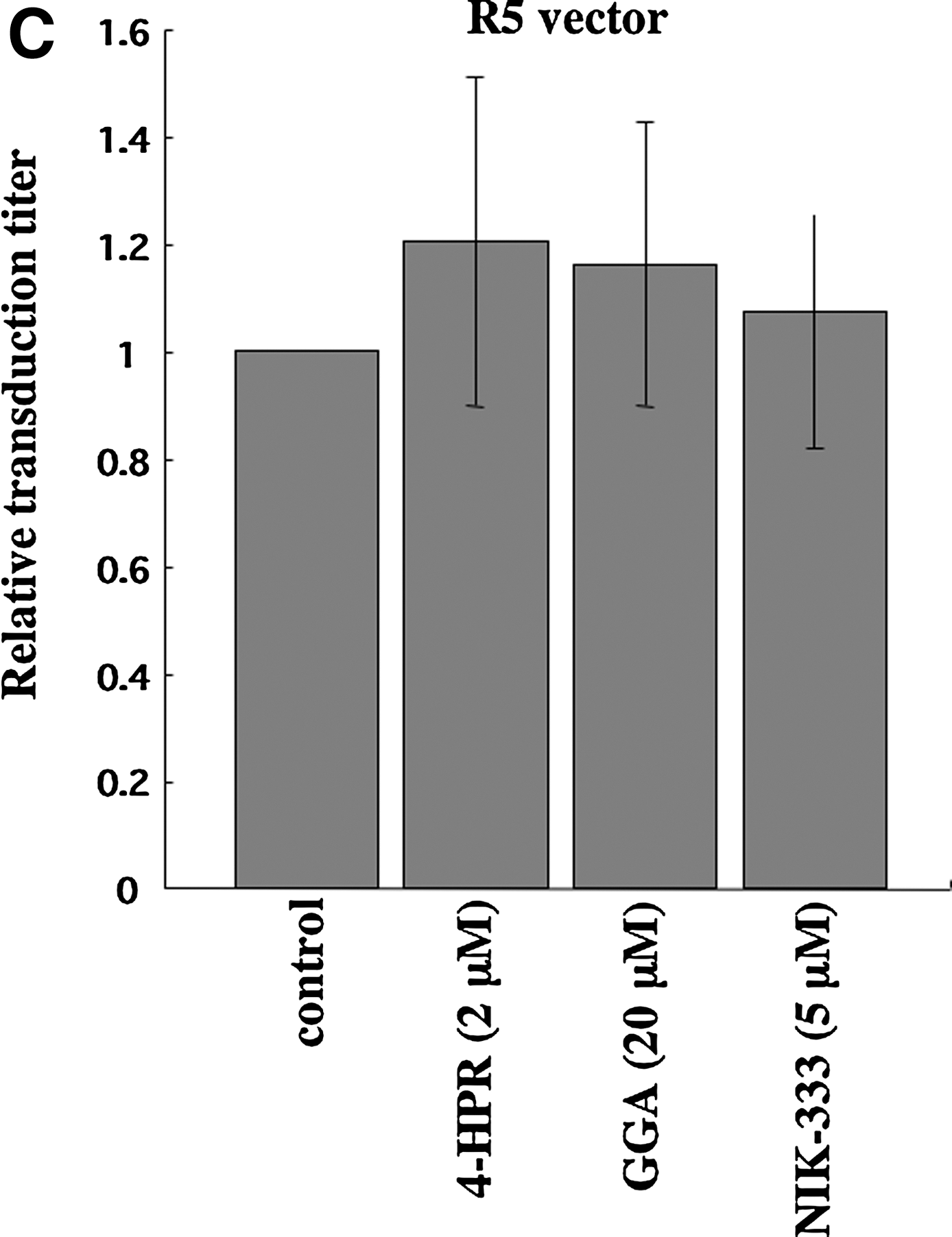
VSV-G-mediated infection is independent of lipid rafts, 4,6 so we assessed whether VSV-G-pseudotyped HIV-1 vector infection is also attenuated by the retinoid analogs. VSV-G-pseudotyped HIV-1 vector infection was not significantly affected by the retinoid analogs (Fig. 2B). Similarly, infection by HIV-1 vector pseudotyped with the Env protein of the CCR5-tropic JRFL strain was not inhibited by the retinoid analogs (Fig. 2C). These results indicate that the retinoid analogs specifically suppress CXCR4-tropic HIV-1 Env-mediated infection but not VSV-G- and CCR5-tropic HIV-1 Env-mediated infection and that the retinoid analogs inhibit CXCR4-tropic HIV-1 infection by a mechanism other than a cytopathic effect.

Retinoid analogs inhibit HIV-1 vector infection. Target cells (NP2/CD4/X4, TE671/CD4, and HeLa/CD4 cells) were either left untreated or pretreated with the retinoid analogs, 4-HPR, GGA, and NIK-333, for 2 days. The cells were then inoculated with the HXB2 Env-
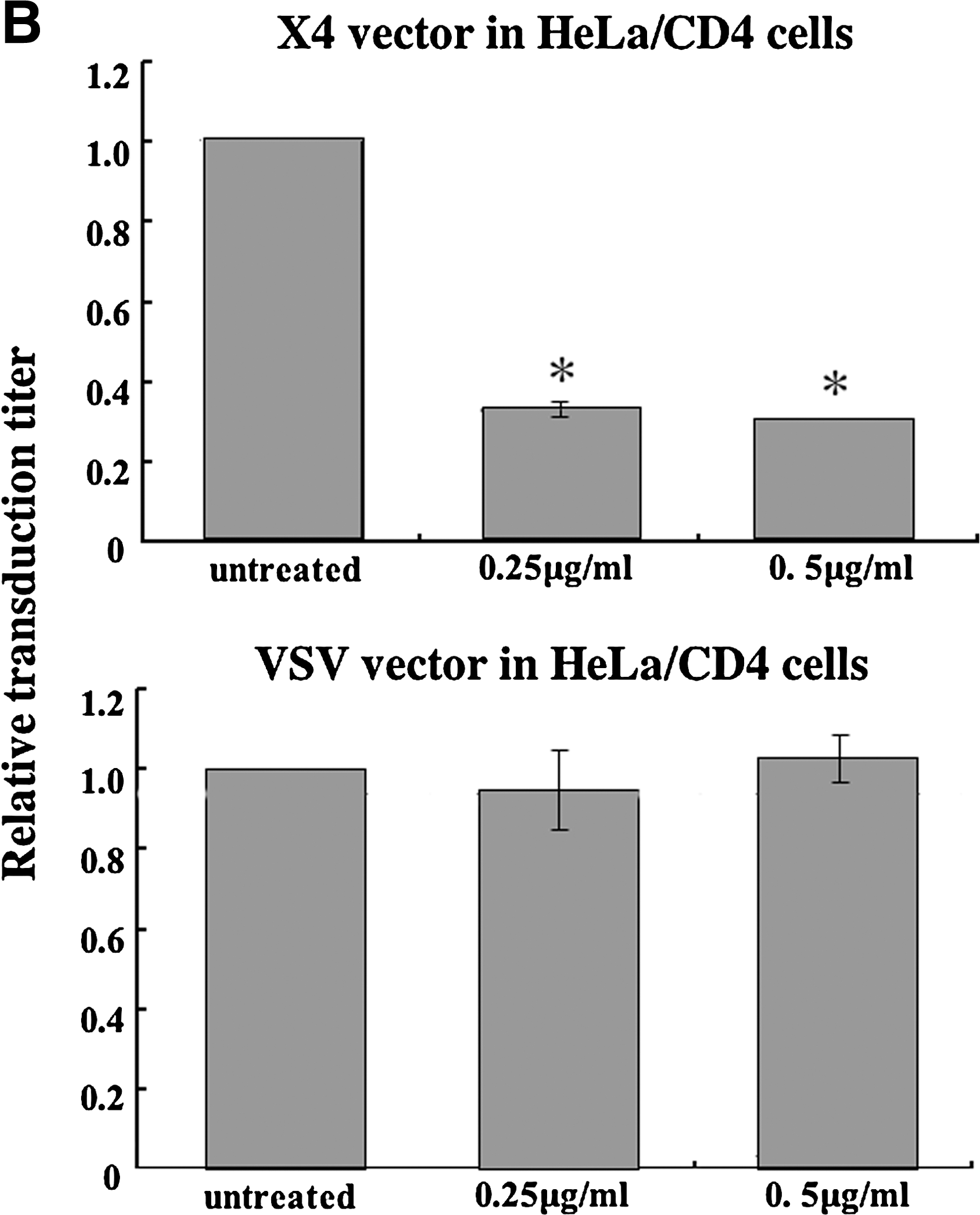
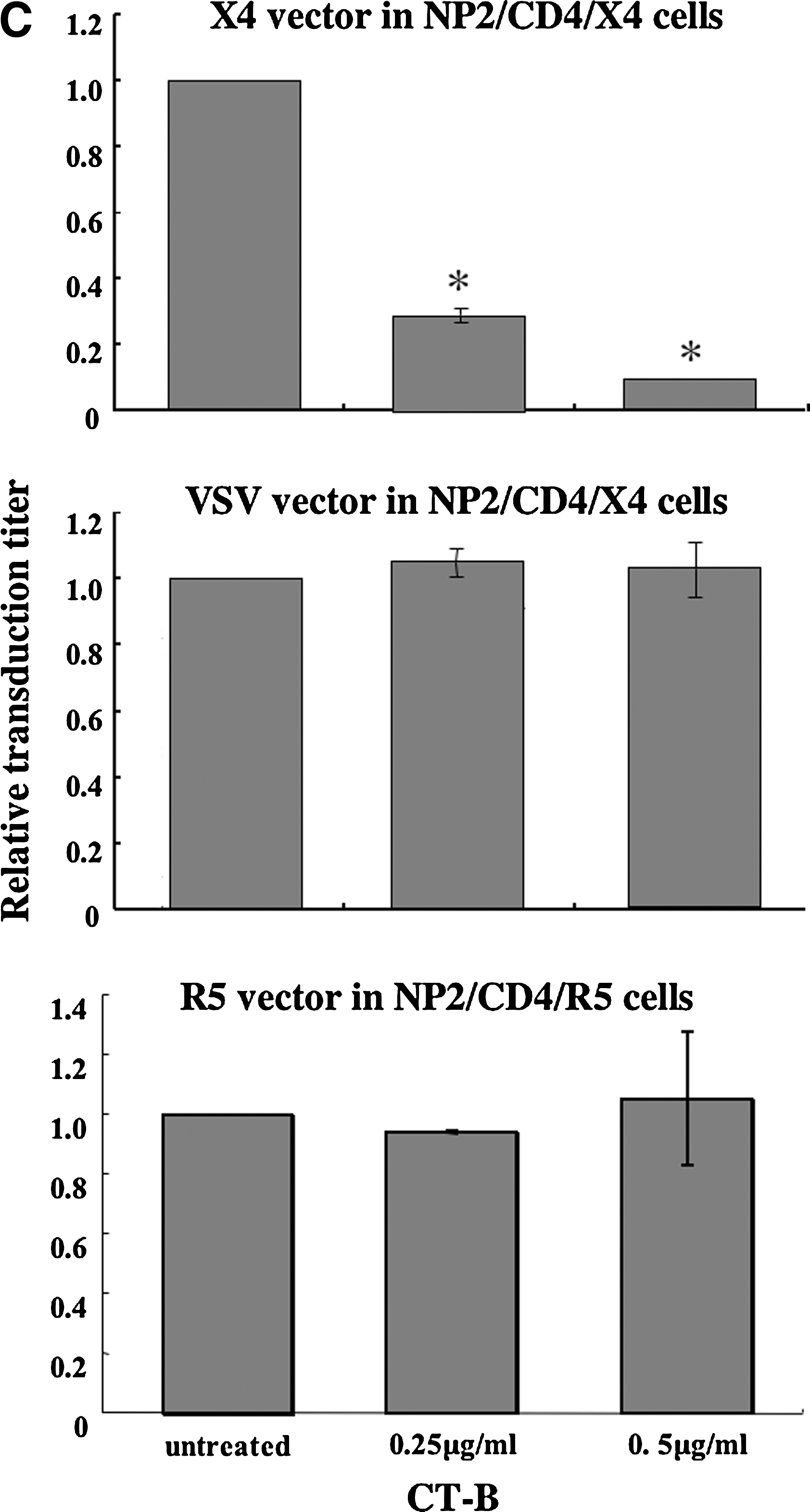
We next assessed whether CT-B inhibits HIV-1 vector infection. CD4-expressing TE671 (TE671/CD4), HeLa/CD4, NP2/CD4/X4, and NP2/CD4/R5 cells were pretreated with CT-B for 24 h and then inoculated with HXB2 Env- or JRFL Env-bearing HIV-1 vector in the absence of CT-B. CT-B significantly attenuated CXCR4-tropic Env-mediated infection but not VSV-G-pseudotyped HIV-1 vector infection in TE671/CD4 (Fig. 3A), HeLa/CD4 (Fig. 3B), and NP2/CD4/X4 cells (Fig. 3C). However, CT-B did not inhibit CCR5-tropic Env-mediated infection in NP2/CD4/R5 cells (Fig. 3C). If CT-B inhibited cell growth, this toxin should also suppress VSV or CCR5-tropic vector infection; however, CT-B did not affect cell growth as analyzed by microscopic observation. These results indicate that CT-B specifically suppresses CXCR4-tropic HIV-1 infection by a mechanism other than cell growth inhibition.
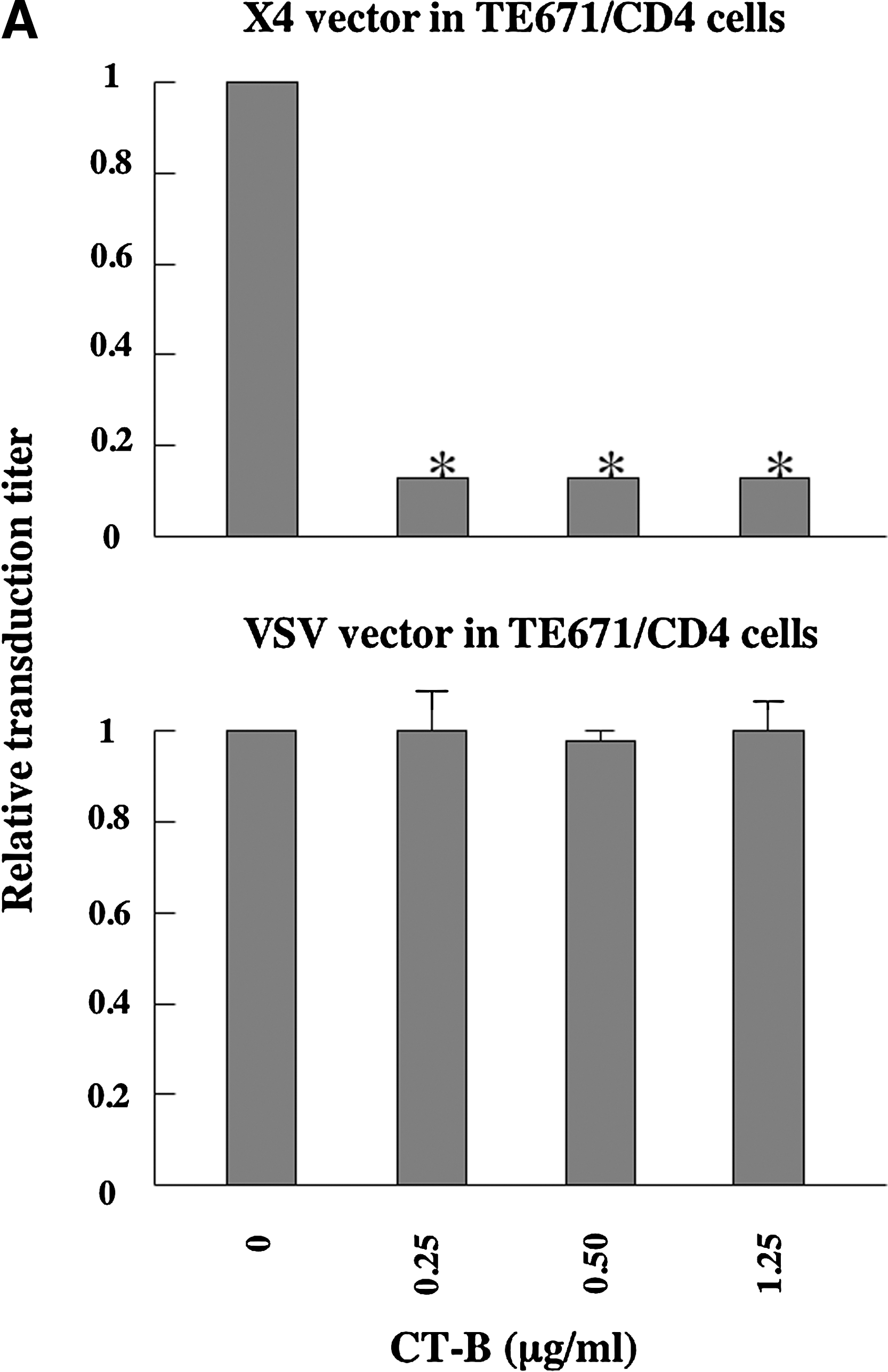
Cholera toxin B (CT-B) inhibits HIV-1 vector infection. TE671/CD4
Additionally, we assessed whether a plant sterol, stigmasterol, inhibits HIV-1 vector infection. The target cells were pretreated with stigmasterol (80 μg/ml) for 24 h. The transduction efficiency of the HIV-1 vector was not affected by the treatment (data not shown).
Retinoid analogs inhibit CXCR4 cell surface expression
As the acyclic retinoid analogs inhibited CXCR4-tropic HIV-1 vector infection, we next assessed whether these retinoid analogs suppressed cell surface expression of the HIV-1 infection receptors, CD4, CXCR4, and CCR5. 4-HPR did not affect CD4 cell surface expression in HeLa/CD4 cells (Fig. 4A). GGA and NIK-333 treatment elevated CD4 expression, though the acyclic retinoid analogs inhibited CXCR4-tropic HIV-1 vector infection. In contrast, all of these retinoid analogs reduced cell surface CXCR4 expression. Similar results were observed in NP2/CD4/X4 cells, in which CXCR4 is artificially expressed (data not shown). Furthermore, these retinoid analogs did not affect CCR5 expression (Fig. 4B). These results suggest that the retinoid analogs inhibit CXCR4-tropic HIV-1 infection by suppressing CXCR4 cell surface expression.
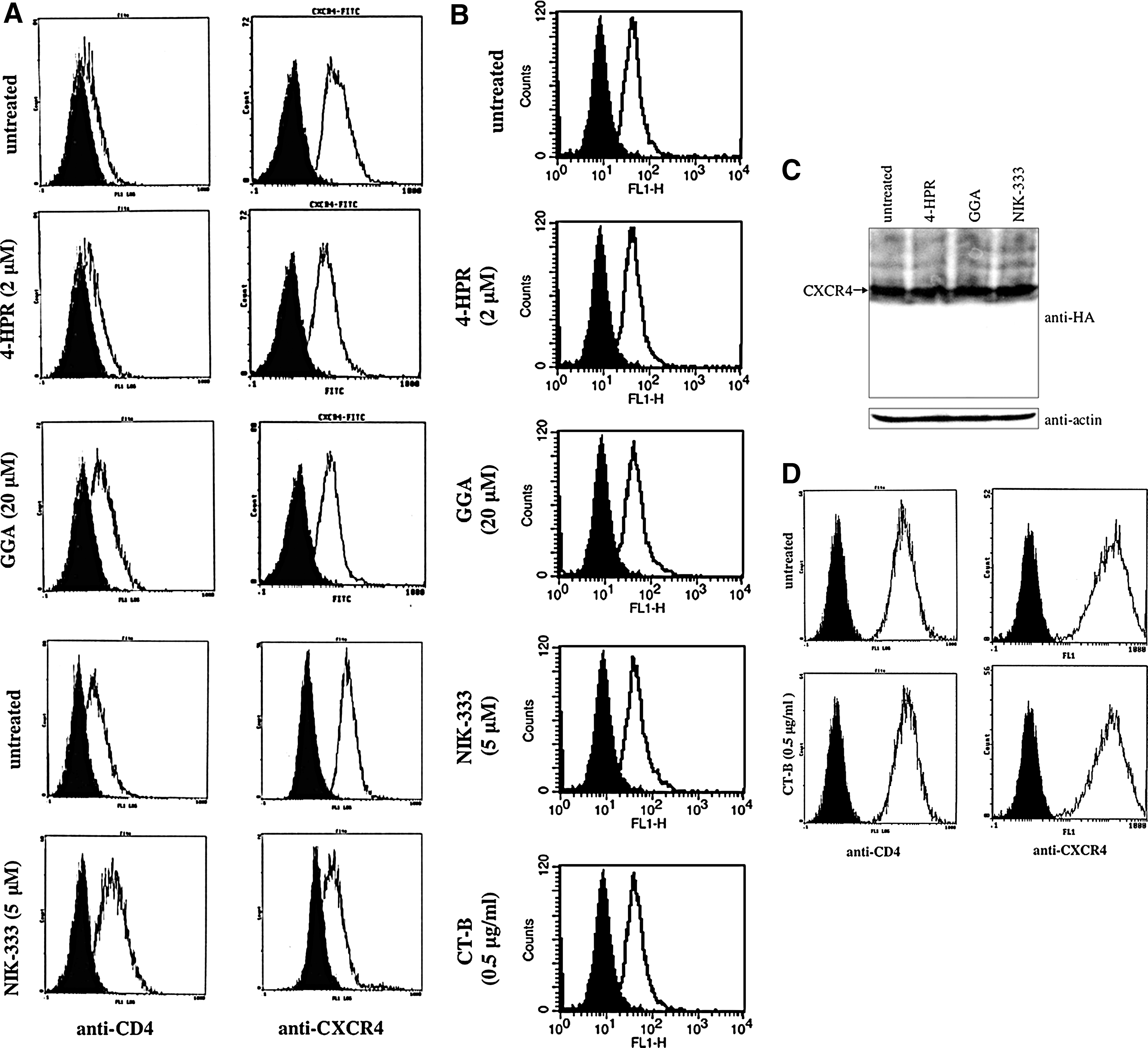
Retinoid analogs inhibit cell surface expression of CXCR4. Cell surface expression of CD4 and CXCR4 in retinoid analog-treated HeLa/CD4 cells
When NP2 cells expressing C-terminally HA-tagged CXCR4 were treated with the retinoid analogs, expression levels of the HA-tagged CXCR4 were not altered, analyzed by Western immunoblotting using an anti-HA antibody (Fig. 4C). This result suggests that the retinoid analogs inhibit the trafficking of CXCR4 to the cell surface, but do not inhibit CXCR4 expression.
CT-B also inhibited CXCR4-tropic HIV-1 vector infection but not CCR5-tropic HIV-1 vector infection; however, CT-B did not affect cell surface expression of CCR5 (Fig. 4B), CXCR4, or CD4 (Fig. 4C). These results indicate that CT-B inhibits CXCR4-tropic infection by a mechanism other than suppression of CXCR4 expression.
Retinoid analogs and CT-B do not affect HIV-1 particle binding to host cells
We analyzed the effects of the retinoid analogs and CT-B on CXCR4-tropic HIV-1 vector particle binding to the target cells by p24 ELISA. The amount of p24 protein bound to CD4-expressing HeLa cells was higher than that bound to CD4-negative HeLa cells, indicating that vector particle binding is CD4-dependent (Fig. 5A). None of the retinoid analogs (Fig. 5B) or CT-B (Fig. 5C) affected HIV-1 vector particle binding to the CD4-expressing target cells. These results show that the retinoid analogs and CT-B inhibit CXCR4-tropic HIV-1 infection by a mechanism other than suppression of CD4-dependent virion binding to target cells.
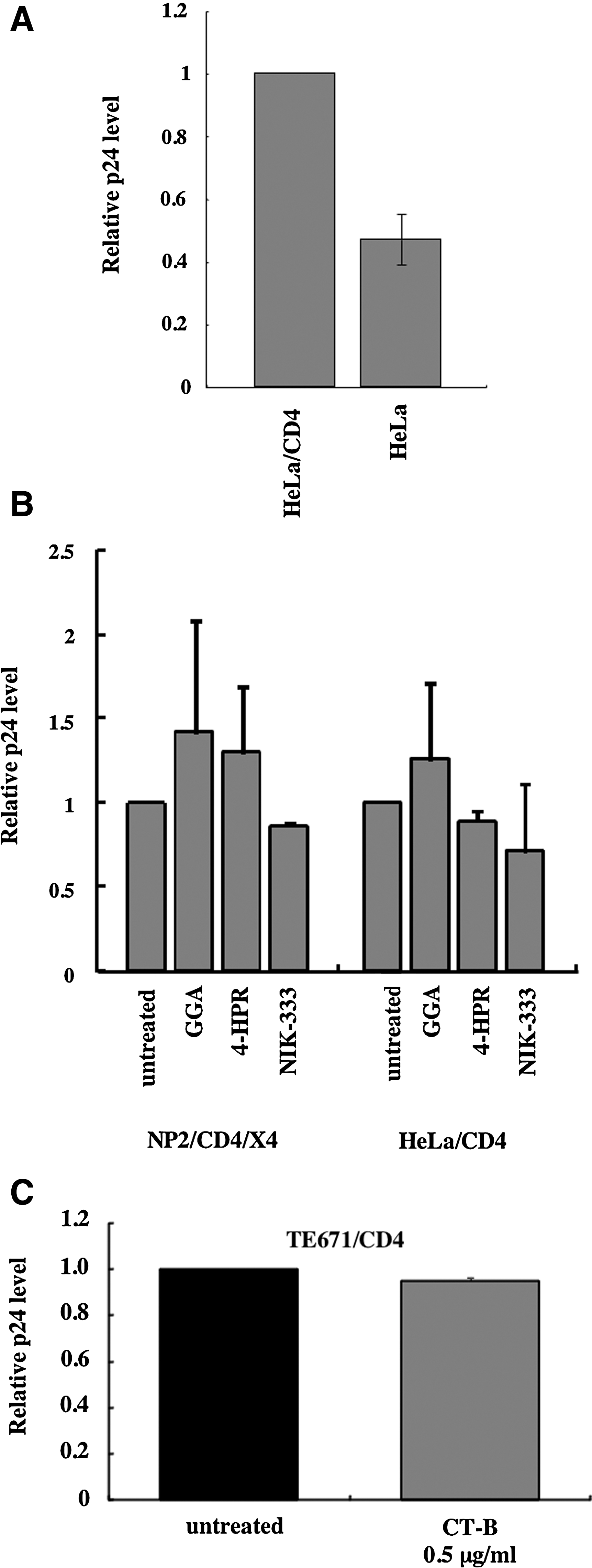
Retinoid analogs and CT-B do not affect HIV-1 vector particle binding to target cells. HeLa/CD4 and HeLa cells were incubated with the HXB2 Env-containing HIV-1 vector particles at 4°C for 1 h and then washed with phosphate buffered saline (PBS)
Retinoid analogs and CT-B inhibit membrane fusion activity of HIV-1 Env protein
To assess whether the retinoid analogs or CT-B inhibit HIV-1 Env-mediated membrane fusion activity, we analyzed the effects of these agents on HIV-1 Env-induced syncytium formation. HEK293T cells transfected with the plasmid encoding the HIV-1 HXB2 Env and Tat proteins were cocultured with MAGIC5 cells for 24 h, and β-galactosidase activity was measured in the cell lysates. The retinoid analogs (Fig. 6A) and CT-B (Fig. 6B) suppressed syncytium formation. Direct inhibition of the HIV-1 Env-mediated membrane fusion reaction by these factors would suppress both CXCR4- and CCR5-tropic HIV-1 infections; however, the factors did not affect CCR5-tropic HIV-1 infection (Fig. 2C). Taken together, these results support the hypothesis that retinoid analogs inhibit CXCR4-tropic HIV-1 infection by attenuating CXCR4 expression, although CT-B may affect the HIV-1 entry process between vector particle binding to target cells and membrane fusion.
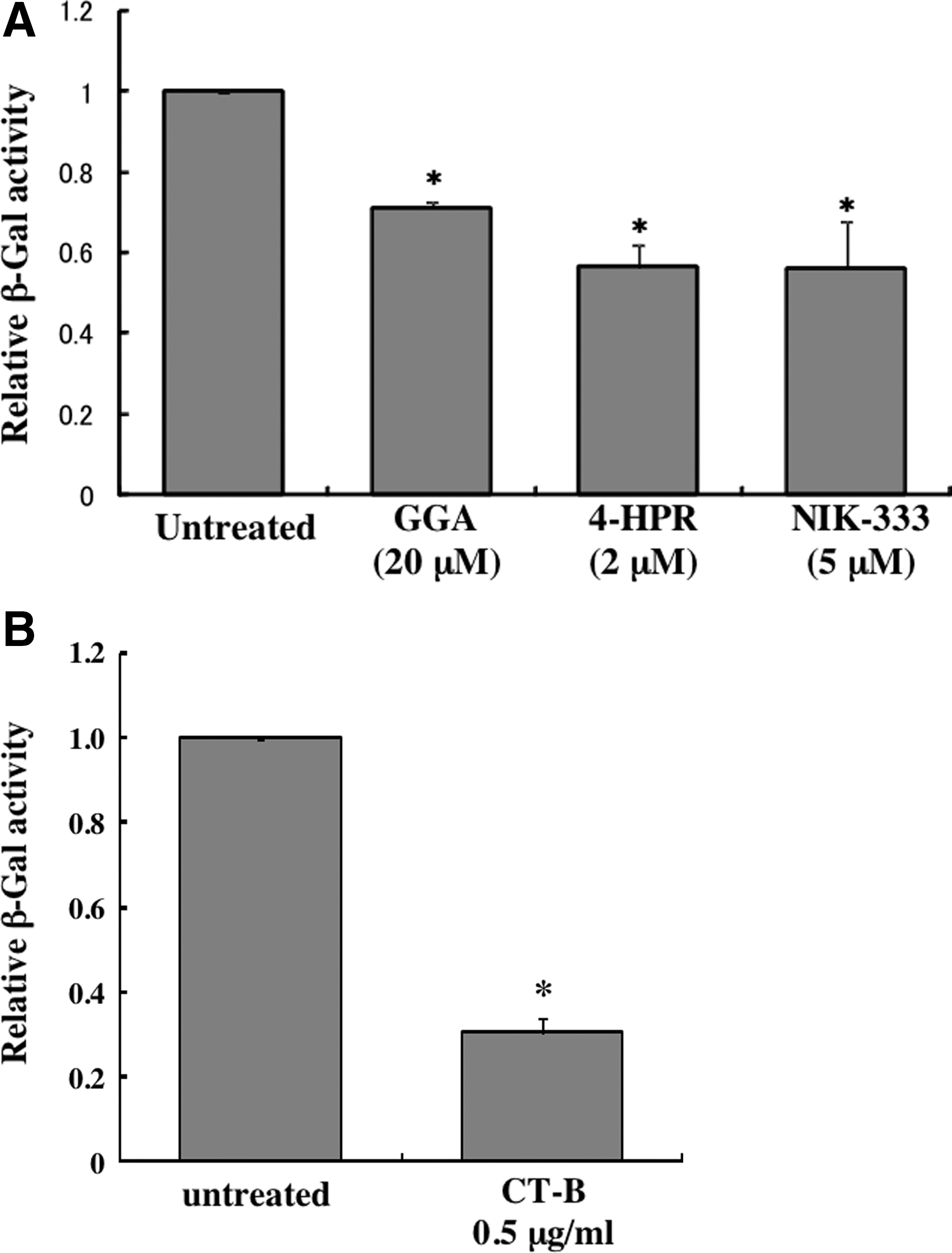
Retinoid analogs and CT-B inhibit CXCR4-tropic HIV-1 Env-induced syncytium formation. Cell fusion activity of the HXB2 HIV-1 Env protein was measured in untreated and retinoid analog-
Discussion
HAART has dramatically reduced the mortality and morbidity of HIV-1-infected patients in developed countries. However, due to the high cost of HAART, this therapy is limited in developing countries. In addition, HIV-1 variants that are resistant to HAART have emerged. Therefore, development of novel low-cost drugs that inhibit HIV-1 replication is essential.
In this study, we found that the acyclic retinoid analogs, GGA and NIK-333, suppress CXCR4-tropic HIV-1 vector infection similarly to 4-HPR. 23 Additionally, retinoids repress expression of the HIV-1 promoter, 38 –40 suggesting that retinoid analogs are possible candidates for a novel anti-HIV-1 therapy. Many reports indicate that vitamin A (retinol) supplementation reduces the mortality of HIV-1-infected patients. 41 –44 NIK333, a synthetic acyclic retinoid, is orally effective against liver cancer without severe side effects. 28 GGA also suppresses HIV-1 vector infection and is present in medicinal herbs. These results suggest that oral intake of a natural acyclic retinoid analog may be novel low-cost therapy against AIDS.
We also found that CT-B efficiently suppresses CXCR4-tropic HIV-1 vector infection. Gauthier and Tremblay have shown that CT-B does not inhibit HIV-1 infection, although the concentration of CT-B (10 ng/ml) used in their study was too low to inhibit HIV-1 infection. 31 Similar to our results with CT-B, pertussis toxin B subunit also inhibits HIV-1 infection. 45 –47 Although the receptor for pertussis toxin B oligomer has not yet been identified, the receptor appears to belong to a class of sialylated glycoproteins, with likely candidates being a 43-kDa protein 48 and CD11b/CD18 integrin. 49 Because the CT-B receptor GM1 is not the pertussis toxin B subunit receptor, the mechanisms by which these bacterial toxin B subunits inhibit HIV-1 infection appear to be different.
One route of HIV-1 transmission is through anal sex. As such, if gut bacteria that secrete nontoxic CT-B are present in the rectum, HIV-1 transmission through this route may be suppressed. Gut bacteria genetically engineered to express CT-B may be a useful novel low-cost strategy to prevent HIV-1 transmission through anal sex.
Use of these factors in vivo, however, should be approached cautiously. First, our study suggests that the acyclic retinoid analogs modulate cell surface expression of CD4 and CXCR4. Second, CT-B is used as an adjuvant for vaccination. 50,51 Therefore, these agents may induce unexpected effects in vivo via activation or perturbation of the human immune system. Further study is required to address this issue.
Other retinoid analogs have been reported to reduce cell surface expression of CXCR4, 52,53 similar to the retinoid analogs used in this study. This down-regulation of CXCR4 expression is one of the mechanisms by which retinoid analogs inhibit CXCR4-tropic HIV-1 infection. HIV-1 infection is suppressed and influenza virus infection is elevated by 4-HPR through activation of endocytosis 23 without suppression of CXCR4 expression. In this study, VSV-G-mediated infection, which occurs via the endosomes, was not affected by the retinoid analogs. Further study is needed to understand the mechanism of HIV-1 infection inhibition by the retinoid analogs.
The retinoid analogs inhibited CXCR4 expression, while CT-B did not, suggesting that the mechanism of HIV-1 infection inhibition by CT-B differs from that by the retinoid analogs. Interestingly, CT-B inhibited CXCR4-tropic HIV-1 infection but not CCR5-tropic infection. Thus, CT-B may inhibit CXCR4-tropic HIV-1 entry at some point between virion binding to host cells and membrane fusion. CD4 and CCR5, but not CXCR4, 4 –6 localize to lipid raft microdomains and constitutively interact. 54,55 It has been reported that CCR5-tropic HIV-1 infection is not dependent upon raft localization of CD4 and CCR5. 56 These results, together with our findings, suggest that CT-B inhibits the HIV-1 Env-induced interaction of CD4 and CXCR4 in lipid rafts and that raft microdomains are differentially involved in CXCR4- and CCR5-tropic HIV-1 infections. CT-B may have no effect on CCR5-tropic HIV-1 infection, because CD4 and CCR5 constitutively interact without binding HIV-1 Env. Recruitment of CXCR4 to CD4-containing raft microdomains by HIV-1 Env, however, has been observed in studies using CT-B as the raft marker. 5,6 Further study is required to understand the mechanism by which CT-B inhibits HIV-1 infection.
The plant sterol stigmasterol did not suppress HIV-1 vector infection. Our group previously reported that MβCD inhibits HIV-1 vector infection and that the addition of cholesterol to the MβCD-treated cells at 50 μg/ml for 30 min recovers infection, suggesting that cholesterol is incorporated into the cell membrane by the addition of cholesterol. 4 Therefore, treatment of cells with stigmasterol at 80 μg/ml for 24 h likely induces uptake of the plant sterol to the cell membrane. These results indicate that this plant sterol does not affect HIV-1 infection. Similar to mammalian cells, plant cells also have lipid raft microdomains in their membranes, 57 and these raft domains are enriched with plant sterols. Therefore, even upon replacement of cholesterol with stigmasterol in mammalian cells, the lipid raft structure is maintained, and HIV-1 infection remains unaffected.
In summary, the acyclic retinoid analogs, GGA and NIK-333, as well as CT-B, efficiently suppress HIV-1 vector infection. Another retinoid analog, 4-HPR, inhibits HIV-1 infection 23 but induces a vitamin A-deficiency syndrome. In contrast, NIK-333 induces no clinical side effects in patients with liver cancer. 28,29 This study suggests that NIK-333 can be used as a novel anti-HIV-1 agent without severe side effects. Additionally, CT-B inhibits CXCR4-tropic, but not CCR5-tropic.
HIV-1 infection, suggesting that host cell lipid raft microdomains are differentially involved in CXCR4- and CCR5-tropic HIV-1 infections.
Footnotes
Acknowledgments
We thank Dr. Y. Yokomaku for the HXB2 and JRFL Env expression plasmids and Dr. H. Hoshino for the NP2/CD4/X4 and NP2/CD4/R5 cells. We also thank Ms. Y. Kobayashi and Ms. F. Tsujita for assistance with laboratory work. This study was supported by the Japan Society for the Promotion of Science (JSPS) (No. 09J07637), a Health Science Research Grant from the Ministry of Health, Labor, and Welfare of Japan, and Kowa Company, Ltd. H. Kamiyama is a special research fellow of JSPS.
Author Disclosure Statement
No competing financial interests exist.