
Abstract
To understand the molecular epidemiology of HIV-1 infection in Iran, we conducted the first study to analyze the genome sequence of Iranian HIV-1 isolates. For this cross-sectional study, we enrolled 10 HIV-1-infected individuals associated with injection drug use from Tehran, Shiraz, and Kermanshah. Near full-length genome sequences obtained from their plasma samples were used for phylogenetic tree and similarity plotting analyses. Among 10 isolates, nine were clearly identified as CRF35_AD and the remaining one as CRF01_AE. Interestingly, five of our Iranian CRF35_AD isolates made two clusters with 10 Afghan CRF35_AD isolates in a phylogenetic tree, indicating epidemiological connections among injection drug users in Iran and Afghanistan. In contrast, our CRF01_AE isolate had no genetic relationship with any other CRF01_AE isolates worldwide, even from Afghanistan. This study provides the first genomic evidence of HIV-1 CRF35_AD predominance and CRF01_AE infection among individuals associated with injection drug use in Iran.
A
In Iran, previous molecular epidemiological analyses of HIV-1 gag and env gene segments had found that the predominant strain circulating among injection drug users (IDUs) was subtype A related to African Ugandan/Kenyan sub-Saharan isolates. 3 –5 Three recent studies in Iran, one on pol gene segments 6 and two on pol and env gene segments, 7,8 reported the circulation of subtype A/D recombinant strain and the predominance of CRF35_AD, respectively. However, no study to date has reported Iranian HIV-1 subtypes based on viral genome sequences. To better understand the molecular epidemiology of HIV-1 infection in Iran, we conducted the first study on genome sequence analysis of Iranian HIV-1 isolates.
For this cross-sectional study, which was approved by the Ethics Committees of Medical Sciences Research at the Tehran University of Medical Sciences and the Ministry of Health and Medical Education, Iran, we enrolled 10 individuals newly diagnosed as HIV-1 infected. They visited the counseling centers for behavioral disease of Tehran, Shiraz, or Kermanshah, Iran between 2010 and 2011. Demographic and clinical characteristics of the subjects are shown in Table 1. After subjects gave written informed consent, their peripheral blood was collected into EDTA-containing vacutainer tubes. Aliquots of plasma sample were stored at −70°C until use. Viral RNA was purified and near full-length genome sequences (8,665 base pairs containing gag to nef genes: positions 784 to 9,448 in the HIV-1 HXB2 sequence) were obtained as previously reported. 9 Sense and antisense primers for reverse transcription-polymerase chain reaction (RT-PCR) were F1-Gag and R1-Nef, respectively. Sense and antisense primers for nested PCR were F2-Gag and R2-Nef, respectively. Nucleotide sequences of primers were as follows: F1-Gag, 5′-GCA GCG ACT GGT GAG TAC GCC-3′; R1-Nef, 5′-AGC ATC TGA GGG TTA GCC ACT CC-3′; F2-Gag, 5′-TTT TGA CTA GCG GAG GCT AGA AGG-3′; and R2-Nef, 5′-CCA CAC CTC CCC TGG AAA GTC CC-3′. Genome sequences were determined using 3130 genetic analyzer with SeqScape software v2.5 (Applied Biosystems, Tokyo, Japan). In phylogenetic tree analyses, multiple sequence alignment was performed using the MUSCLE program, and genetic distances were calculated based on the maximum composite likelihood model using MEGA software v5.05. 10 Phylogenetic trees were constructed using the neighbor-joining method with 1,000 bootstrap analyses. In similarity plotting analyses, genome sequences of nine subtypes (A–D, F–H, J, and K) and two CRFs (CRF01_AE and CRF35_AD) were used as references. After realigning the sequence data set, similarity plotting was performed using SimPlot software v3.5.1 with window and step sizes of 300 and 20 nucleotides, respectively. 11 HIV-1 subtype reference sequences were obtained from the HIV Sequence Database at the Los Alamos National Laboratory. 2
M, male; F, female; IDU, injection drug user; NA, not available; HCV, hepatitis C virus; HBV, hepatitis B virus; +, positive; −, negative.
Of the 10 HIV-1-infected individuals enrolled in this study, four were from Tehran, three from Shiraz, and three from Kermanshah with a median age of 26 years old (Table 1). Six (60%) were males whose transmission route was injection drug use, and four (40%) were females whose transmission route was heterosexual contact with their husbands using injected drugs. Thus, all the HIV-1 infections were associated with injection drug use. Three cases were identified as coinfected with hepatitis C virus (43%, 3/7), whereas no cases were coinfected with hepatitis B virus (0%, 0/7). All subjects had never received antiretroviral therapy. From their plasma samples, we successfully obtained 10 near full-length HIV-1 genome sequences. To determine the HIV-1 genotypes of these 10 sequences, we first constructed a phylogenetic tree with HIV-1 references containing nine subtypes and all CRFs available in public databases (CRF01 to 51 except for 30, 41, and 50). As shown in the summarized tree (Fig. 1), nine of our isolates made a cluster with CRF35_AD references and the remaining one with CRF01_AE references. To obtain further evidence for HIV-1 subtyping, we performed similarity plotting analyses. As shown in Fig. 2 (left side), all nine CRF35_AD candidates showed the highest similarity to the CRF35_AD reference all over the genome. A tiny peak of highest similarity to the subtype A1 reference was observed near the beginning of plots in four cases: 10IR.THR01F, 11IR.KSH24F, 11R.KSH31F, and 10IR.THR41F (Fig. 2, left side). However, there was no discrepancy because the gag gene of CRF35_AD was derived from subtype A1. The remaining isolate, 10IR.THR48F, clearly showed the highest similarity to the CRF01_AE reference all over the genome (Fig. 2, right side). Given all the data, we can conclude that nine of our isolates are HIV-1 CRF35_AD and one is HIV-1 CRF01_AE.

Phylogenetic tree analysis of Iranian HIV-1 isolates. The tree was constructed with near full-length HIV-1 genome sequences. Bootstrap values greater than 80% are shown at the tree nodes. Our 10 isolates are represented by filled circles, and subtype reference isolates are represented by their subtype and name. The scale bar represents nucleotide substitutions per site. HIV-1 group O isolate, ANT70, was used as the outgroup.
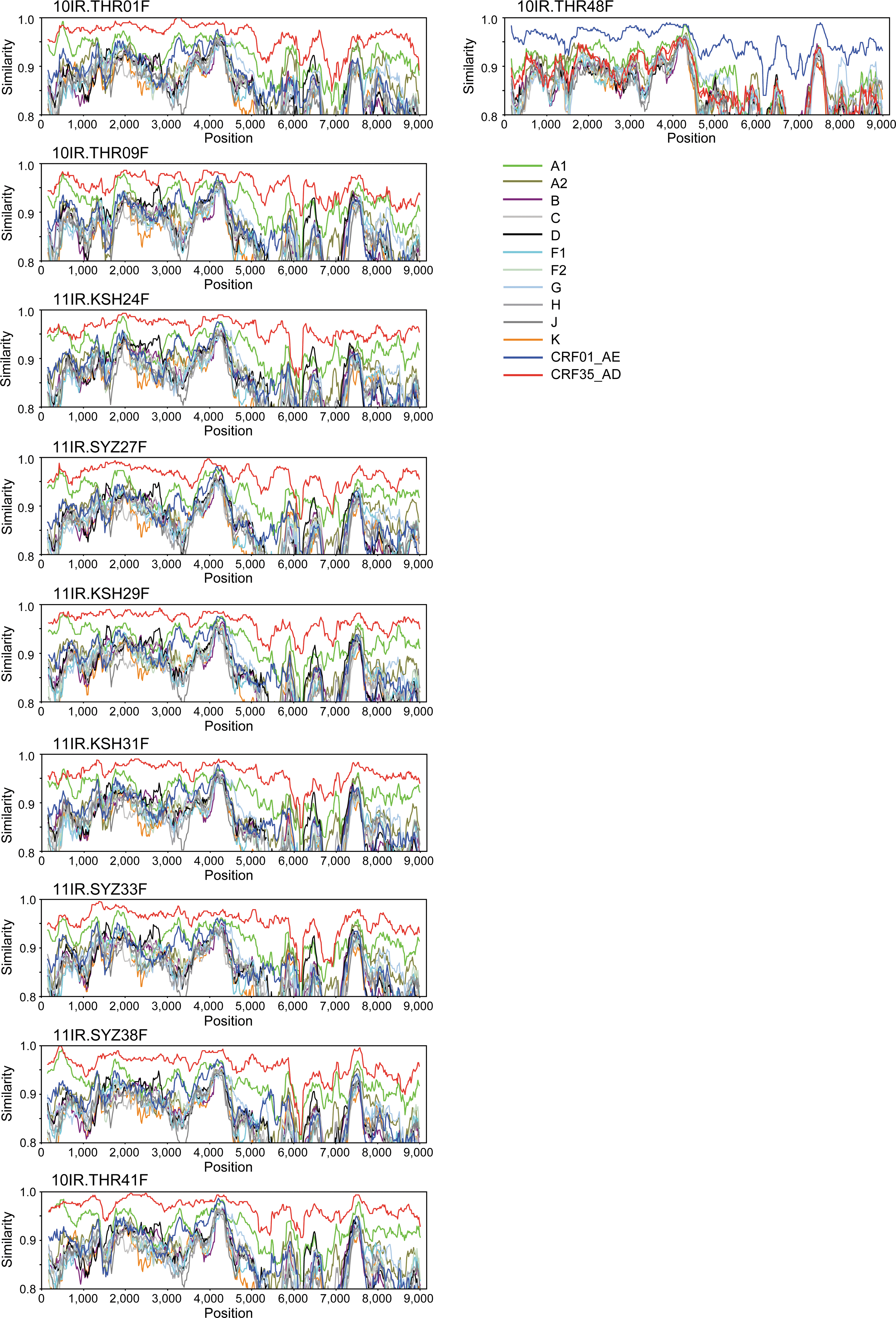
Similarity plotting analyses of Iranian HIV-1 isolates. Similarity plotting was performed with window and step sizes of 300 and 20 nucleotides, respectively. The color code for each consensus sequence of A1, A2, B, C, D, F1, F2, G, H, J, K, CRF01_AE, and CRF35_AD is shown below the 10IR.THR48F data.
To better understand the molecular epidemiology of HIV-1 CRF35_AD infection, we constructed another phylogenetic tree by adding 14 CRF35_AD genome sequences reported from a neighboring country, Afghanistan. 12,13 Interestingly, two clusters were identified among the CRF35_AD isolates (Fig. 3A). The first major cluster comprised 12 isolates (52%, 12/23): nine Afghan isolates (eight from Herat and one from Kabul) and three Iranian isolates (two from Tehran and one from Kermanshah). The second minor cluster comprised three isolates (13%, 3/23): two from Shiraz, Iran, and one from Kabul, Afghanistan (Fig. 3A and B). Finally, to track the origin of our single HIV-1 CRF01_AE isolate, we constructed another phylogenetic tree using 155 CRF01_AE genome sequences reported from around the world. Although we identified 22 clusters in the tree, our isolate 10IR.THR48F was not involved in any of these clusters (Fig. 3C). Of note, in contrast to the CRF35_AD case, our CRF01_AE isolate had no genetic relationship with a CRF01_AE isolate from Afghanistan (Fig. 3C).

Phylogenetic tree analyses of HIV-1 CRF35_AD and CRF01_AE isolates.
The work presented here is the first report on the genome sequence analysis of HIV-1 isolates in Iran. Near full-length genome sequences were successfully obtained in 10 HIV-1-infected individuals associated with injection drug use. Among the 10 HIV-1 isolates, nine were clearly identified as CRF35_AD and the remaining one as CRF01_AE through phylogenetic tree and similarity plotting analyses. The predominance of CRF35_AD shown in this study is consistent with two recent reports from Iran, in which pol and env gene segments were used for HIV-1 subtyping. 7,8 Before the discovery of CRF35_AD, 12 three studies on gag and env gene segments had reported that the predominant HIV-1 circulating among IDUs in Iran was subtype A. 3 –5 However, in the presence of CRF35_AD references, their subtype A isolates made a cluster with the CRF35_AD references rather than subtype A references in phylogenetic trees (data not shown), suggesting the substantial history of CRF35_AD predominance among IDUs in Iran. In this study, we identified one CRF01_AE-infected case, an IDU from Tehran (subject #48 in Table 1). This finding is noteworthy because two CRF01_AE-infected cases were recently reported in a study on pol gene segments (5%, 2/42); one case was an IDU and one case had an unknown transmission route. 6 Therefore, we also need to pay careful attention to the spread of CRF01_AE in Iran. To our surprise, after adding 14 CRF35_AD isolates from IDUs in Afghanistan 12,13 to our phylogenetic tree analysis, more than half of Iranian and Afghan CRF35_AD isolates (52%, 12/23) gathered into a single cluster (cluster 1 in Fig. 3A). A second cluster also contained both Iranian and Afghan isolates (cluster 2 in Fig. 3A). The data clearly indicate epidemiological connections among IDUs in Iran and Afghanistan. In contrast, we found no genetic relationship between two CRF01_AE isolates from Iran and Afghanistan (Fig. 3C). However, the absence of an epidemiological connection between the CRF01_AE infections could be explained by a distinct difference in transmission routes; our Iranian case was an injection drug user (subject #48 in Table 1) and the Afghan case was a commercial sex worker. 13
The epidemiological connection among IDUs in Iran and Afghanistan shown in this study can be explained by drug trafficking and/or immigration. Afghanistan was the world largest opium-producing country in 2010, responsible for 74% of global opium production, and Iran was identified as a major route for drug trafficking from Afghanistan to Europe. 14 The data allow us to speculate that HIV transmission among IDUs occurs along the route of drug trafficking from Afghanistan to Iran. Although Iran has been the world's best country in contributing to global opiate seizures (89% in 2009), 14 their efforts in blocking the route should be continued to also prevent the spread of HIV in the area. On the other hand, Iran has received the largest number of Afghan refugees. Therefore, we cannot reject the possibility that the expansion of CRF35_AD in Iran is due in part to Afghan IDUs who immigrated to the county. However, further detailed studies are needed to examine this possibility.
In conclusion, we provide here the first genomic evidence of CRF35_AD predominance and CRF01_AE infection among individuals associated with injection drug use in Iran. According to the latest report from Iran's Ministry of Health and Medical Education, injected drug use remains the primary transmission route of HIV infection in the country (69.8% in September 2011). 15 Therefore, current harm-reduction programs for IDUs in Iran need to be strengthened to prevent further HIV diffusion among IDUs and from IDUs to other populations.
Sequence Data
Nucleotide sequences obtained in this study were deposited in the DNA Databank of Japan under accession numbers AB703607 to AB703616.
Footnotes
Acknowledgments
We thank the staff members at the counseling centers for behavioral disease (triangular clinics) of Tehran, Shiraz, and Kermanshah for their help in collecting clinical samples. We also thank the staff at the Hepatitis and AIDS Department and Virology Department of the Pasteur Institute of Iran for their technical support. We are grateful to the personnel at the Clinical Research Center of Nagoya Medical Center for their help in performing experiments. We thank Ms. Claire Baldwin for her help in preparing the manuscript. This study was supported by grants from the World Health Organization, the Ministry of Health and Medical Education, Iran, and the Ministry of Health Labour and Welfare, Japan.
Author Disclosure Statement
No competing financial interests exist.