
Abstract
The presence of infection by human T cell lymphotropic virus type 1 (HTLV-1) in Cuba has been previously documented. However, genetic information on the strains that circulate in the Cuban people remains unknown. The present work constitutes the first study of phylogenetic relationship of HTLV-1 Cuban isolates. Twelve Cuban patients who were diagnosed with HTLV-1 infection and had different clinical manifestations were studied. The 3' LTR sequences were analyzed for the construction of a phylogenetic tree with reference sequences of HTLV-1 of different geographic origins. Phylogenetic analysis of the 3' LTR gene showed that all the Cuban samples clustered in the Transcontinental subgroup of the Cosmopolitan subtype. Phylogenetic analysis suggests multiple introductions of HTLV-1 in Cuba as well as a possible African origin of the samples. The results of the study will reinforce the program of epidemic surveillance of the infection in Cuba.
T
Four main HTLV-1 subtypes have been identified based in phylogenetic analysis of the 3' LTR gene: the Cosmopolitan (a), the Central African (b and d), and the Melanesian (c). The Cosmopolitan group is disseminated around the world and has been divided into five subgroups: Transcontinental (A), Japanese (B), West African/Caribbean (C), North African (D), and Black Peruvian (E). 4 In the Americas, it has been suggested that the Cosmopolitan group was introduced during the post-Columbus migrations due to the African slave trade and some Japanese migrants. 5 In Cuba, the first evidence of HTLV-1 infection was found by serological surveillance carried out in different risk groups where an antibody seroprevalence of 0.037% was reported 6,7 and later in a patient with clinical manifestations of ATLL diagnosed in 1990. 8
Since 1990, a total of 40 individuals have been serologically confirmed as positive for antibodies against HTLV-1 at the AIDS Research Laboratory––the Cuban National Reference Laboratory for Human Retrovirus. Nevertheless, there are no available reports concerning the genetic information of circulating HTLV-1 strains in the Cuban population. The actual research was done in order to determine the genetic diversity of HTLV-1 strains isolated from asymptomatic Cuban individuals and/or from Cuban patients with HTLV-1-associated diseases.
Between January 2010 and December 2011 a total of 12 HTLV-1-infected individuals were investigated and 10 ml EDTA blood sample was taken; this was done after the completion of a written informed consent and the recording of clinical and epidemiological data, including the patient's age, sex, origin, and method of infection (Table 1). The infection source or acquisition mode was determined by the analysis of epidemiological evidence and a serological investigation of family (parents, children, brothers, and sisters) and sexual contacts. All sera samples were screened with an HTLV-1 antibodies immunoassay test (DAVIH HTLV-1, DAVIH Laboratories, Cuba) according of the manufacturer's directions. Positive samples were later confirmed using a Western blot assay (DAVIH-blot HTLV-1, DAVIH Laboratories, Cuba) according to the manufacturer's directions. Positive individuals were classified as asymptomatic carriers, TSP/HAM, and ATLL patients, according the World Health Organization (WHO) directives. 9
F, female; M, male; TSP/HAM, tropical spastic paraparesis/HTLV-1-associated myelopathy; ATLL, adult T cell leukemia/lymphoma.
DNA was extracted from whole blood by midi spin columns from the High Pure Viral Nucleic Acid kit (Roche Diagnostic, IN) following the manufacturer's directions. Samples were subjected to an “in house” nested polymerase chain reaction (PCR) to amplify tax and pol genes. Amplification of the pol region was performed with outer primers SK-110/SK-111-I and inner primers pol 1.1/pol 3.1 and pol 1.2/pol 3.2. 10 Amplification of the tax region was carried out with outer primers SK-43-I/SK-44-I and inner primers SK-43/SK-44. 11 The PCR reactions were performed by use of the Fast Start High Fidelity PCR System (Roche Diagnostics GmbH, Mannheim, Germany). The PCR products were 135 bp for pol and 128 bp for tax amplification.
To perform the phylogenetic analysis, the 3' LTR region was amplified by heminested PCR by using 8200LA/3Vext as the outer primers and 8200LA/3Vint as the inner primers (528 bp, ATK-1 genome positions 8196–8699). 12 Sequencing in both directions was performed using the Genome Lab Dye Terminator Cycle Sequence with Quick Start kit following the manufacturer's directions (Beckman Coulter, Inc., CA) and with the inner primers of the heminested PCR employed. 12 Sequencing products were read on a CEQ 8800 genetic analyzer (Beckman Coulter, Inc., CA). Neighbor-joining (NJ) and maximum likelihood (ML) trees were generated by PAUP 4.0. 13 The TIM+G model was selected as the best model for phylogenetic analysis (alpha parameter=0.7966). The nucleotide model was inferred using Modeltest. 14 The reliability of the NJ trees was assessed by analyzing 1,000 bootstrap replicates. For ML trees, a heuristic search was performed with a subtree pruning regrafting branch swapping algorithm using the NJ tree as the starting material, including its optimized parameters. The likelihood ratio test (RT) method was used to calculate statistical support for the branches. Bootstrap values for the ML analysis were obtained with the PHYML program. 15
Of the seven (58.3%) samples studied a number were from patients with HTLV-1-associated diseases. From them, five (41.6%) have developed HAM/TSP, one patient developed a cutaneous T cell lymphoma, and the other developed uveitis (Table 1).
The 3' LTR region phylogenetic analysis allowed grouping the 12 patient's nucleotide sequences in the Cosmopolitan subtype [Transcontinental (A) subgroup], with a bootstrap value of 73% (p≤0.001 for the ML analysis). The majority of our studied sequences (n=10) were grouped by their homology with sequences coming from French Guyana. The 11CUHT01 sample was grouped inside the Latin American cluster beta. The 11CUHT12 formed a group with sequences coming from French Guyana and Chile (Fig. 1).
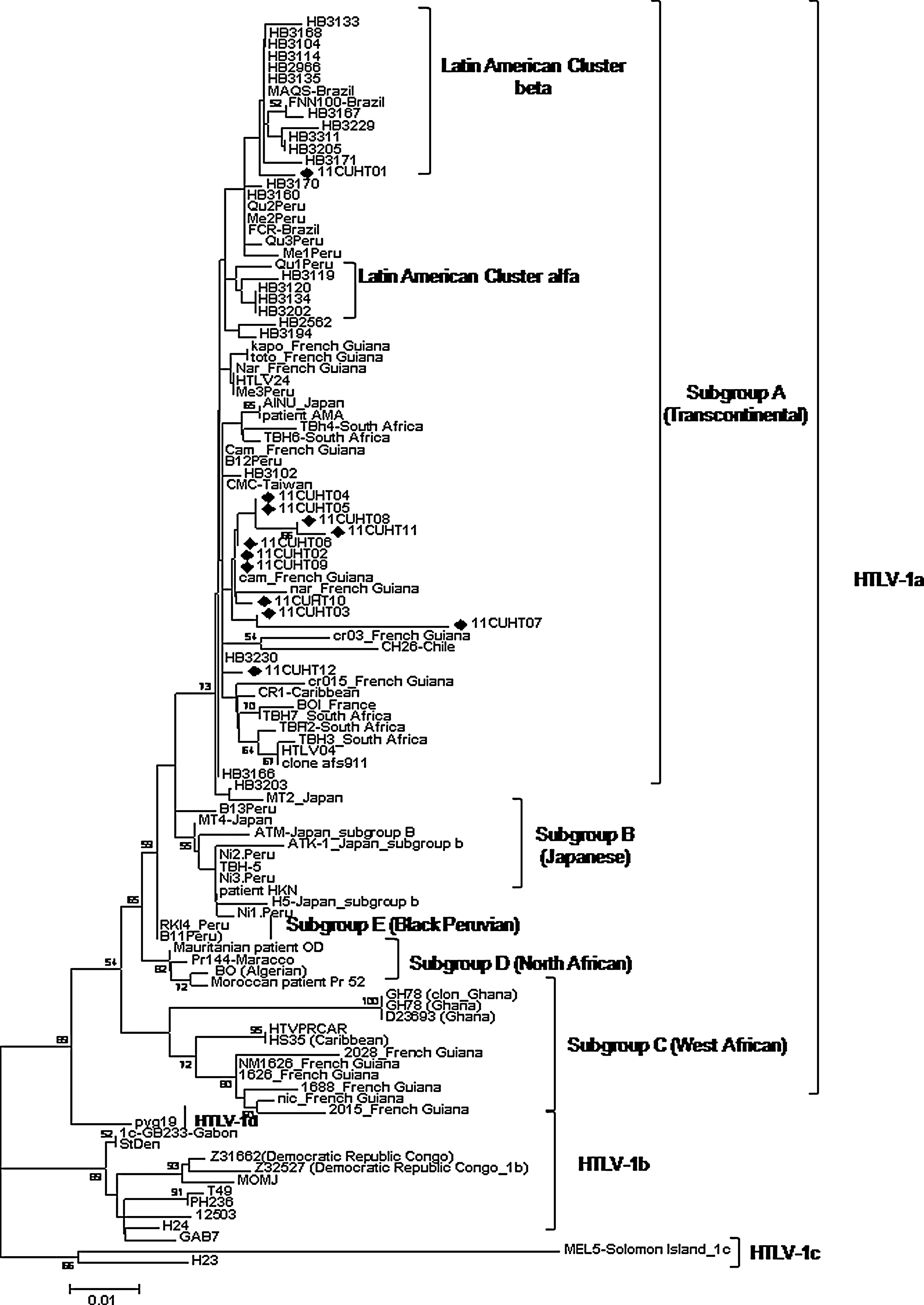
Maximum likelihood (ML) tree of 3' LTR human T cell lymphotropic virus type 1 (HTLV-1) sequences. ML analysis was performed under the TIM+G nucleotide substitution model. All 12 Cuban sequences are indicated by a solid diamond (♦). Numbers on branches indicate the degree of support for each node. MEL5 was used as the outgroup. Geographic origin is given in parentheses.
Although the presence of HTLV-1 in Cuba was already documented, 6 –8 this report is about the first study of circulating HTLV-1 genetic variants in the Cuban seropositive population.
The presence of HTLV-1 subtype A subgroup C in the Caribbean is supported by the introduction of this linage from Western Africa during the slave trade. 4 Nevertheless, circulation of the Transcontinental subgroup has been detected in the area, so it is not surprising that all Cuban strain sequences were found inside this subgroup. Their grouping in different individual clusters suggests that there were multiple introductions of HTLV-1 in Cuba. Nevertheless, it is important to note that sample 11CUHT01, grouped together with sequences from Brazil in the Latin American cluster beta, was isolated from a female patient who has had sexual intercourse with an individual infected in Africa, according to the epidemiological surveillance data. Several phylogenetic studies carried out in some Brazilian populations have also shown an association between the Latin American cluster beta with sequences from African origin. 16,17
Inside the group of samples with sequences similar to the ones from French Guyana, the 11CUTH02 sample is from a patient who infected both 11CUTH03 and 11CUHT11 samples by sexual and vertical transmission, respectively. 18,19 The high degree of homology of the studied sequences in these three samples is evidence of the same genetic variant transmission, in spite of the fact that there is a little distance between them, inside the same phylogenetic cluster.
The 11CUHT06 sample was obtained from a direct descendent of Haitians and he is the father of patients with the 11CUHT05 and CUHT09 samples. This result suggests that one of the possible origins of the HTLV-1 infection in Cuba results from the migration of people from Haiti, a country where TSP is frequent, and TSP has been used as a sentinel disease indicating a high prevalence of HTLV-1 infection. The Afro-Caribbean origin of part of the Cuban population 20 together with molecular evidence of the arrival of HTLV-1 in the Americas from the African population introduced by the European colonizers 21,22 are factors that can explain the origin of HTLV-1 in Cuba.
The 11CUHT12 sample is from a female patient born in Santiago de Cuba province who developed HAM/TSP and it was grouped by homology with nucleotide sequences coming from the Latin American area (French Guyana, Chile, and Brazil). The Santiago de Cuba province, with its Caribbean seacoast, is an area known to be used for the entrance of Haitian immigrants to Cuba, which may reinforce the relationship between this sample and the previously mentioned Caribbean origin sequence samples.
The clinical presentation of HTLV-1 in the Caribbean differs from that observed in Japan and sub-Saharan Africa. In the Caribbean HAM/TSP predominates while in Japan ATLL is more frequent, with 86 new cases of the leukemia and only three patients with the myelopathy per 100,000 inhabitants per year. 23 Our phylogenetic results have epidemiological importance and can have prognostic value, because it has been indicated that individuals infected with HTLV-1 Transcontinental subgroup A may be associated with a higher risk for the development of HAM/TSP compared to individuals infected with HTLV-1 subgroup B. 24
In conclusion, this article shows the presence of the subtype A Transcontinental subgroup A of HTLV-1 in Cuba. Finally, the results obtained will contribute to the epidemiological surveillance program; they will reinforce the studies about the origin of HTLV-1 in Cuba and the detection of patterns of evolution for HTLV-1 circulating genetic variants in the country.
Nucleotide Sequence Accession Numbers
GenBank accession numbers for the sequences reported here are from JX194169–JK194179 and JX871882.
Footnotes
Author Disclosure Statement
No competing financial interests exist.