
Abstract
HIV-1 tropism can be predicted using V3 genotypic algorithms. The performance of these prediction algorithms for non-B subtypes is poorly characterized. Here, we use these genotypic algorithms to predict viral tropism of HIV-1 subtype A, B, C, and D to find apparent sensitivity, specificity, and concordance against a recombinant phenotypic assay, the original Trofile assay. This is a substudy of an epidemiological study (Pfizer A4001064). Plasma samples were selected to represent a large number of DM/X4 and R5 viruses. The HIV-1 env gene V3 loop was genotyped by Sanger sequencing (N=260) or 454 “deep” sequencing (N=280). Sequences were scored with g2p[coreceptor], PSSM X4/R5, PSSM SI/NSI, and PSSM subtype C matrices. Overall, non-B subtypes tropism prediction had similar concordance and apparent sensitivity and specificity as subtype B in predicting Trofile's results in both population sequencing (81.3%, 65.6%, and 90.5% versus 84.2%, 78.5%, and 88.2%) and 454 “deep” sequencing (82.3%, 80.0%, and 83.6% versus 86.8%, 92.0%, and 82.6%) using g2p[coreceptor]. By population sequencing, subtype A had lower sensitivity, whereas subtype D had lower specificity for non-R5 predictions, both in comparison to subtype B. 454 “deep” sequencing improved subtype A sensitivity but not subtype D. Subtype C had greater concordance than subtype B regardless of sequencing methods. In conclusion, genotypic tropism prediction algorithms may be applied to non-B HIV-1 subtypes with caution. Collective analysis of non-B subtypes revealed a performance similar to subtype B, whereas a subtype-specific analysis revealed overestimation (subtype D) or underestimation (subtype A).
Introduction
HIV
MVC received regulatory approval based partially on the MOTIVATE-1 and −2 trials. These trials enrolled subjects predominantly infected with subtype B viruses (94%) and R5 viral tropism as determined by a cell-culture-based phenotypic assay, the original Trofile assay. 4,5 We have previously reported retrospective analysis of the MOTIVATE-1/-2 studies plus the A4001029 and the MERIT studies, which include non-R5 samples using population-based or 454 “deep” sequencing-based methods examining the V3 loop of the HIV-1 env gene. Viral tropism was inferred with bioinformatic algorithms including g2p[coreceptor] 6 or PSSM 7 algorithms. These analyses demonstrated that both Trofile-determined viral tropism (R5, DM for dual mixed, or X4) and virological outcome on MVC could be predicted by sequencing approaches. These approaches also had performance similar to the Enhanced Sensitivity Trofile Assay (ESTA) in the MERIT trial. 8
Some of the currently available tropism prediction algorithms were developed using a single subtype (e.g., PSSM was trained on subtype B 7 or C 9 ) while other algorithms were trained on a mixture of subtypes (e.g., g2p[coreceptor] 6 ). Since only 6% of participants in MVC's clinical trials hosted non-B virus, 4 limited data are available on the specific performance of these genotypic algorithms in predicting HIV-1 tropism in non-B subtypes. A previous study assessed the compatibility of the original Trofile assay with non-B HIV-1 subtypes (Pfizer A4001064, N=736). 10 The purpose of that study was also to estimate the prevalence of R5, DM, and X4 viruses in South Africa and Uganda and to evaluate and optimize the performance of the original Trofile assay for non-B subtypes. These samples had not been previously analyzed by genotypic methods. This study consisted of N=736 subjects: N=286 hosted subtype A HIV-1 by protease/reverse transcriptase sequences, were from Uganda, were treatment naïve or experienced, with 240/286 (84%) R5, 46/286 (16%) non-R5 by the original Trofile assay; N=256 hosted subtype C HIV-1, were from South Africa, were treatment experienced, with 179/256 (70%) R5, 77/256 (30%) non-R5; N=194 hosted subtype D HIV-1, were from Uganda, were treatment naïve or experienced, with 144/194 (74%) R5, 50/194 (26%) non-R5 viruses.
The objective of the current study was to determine the concordance, sensitivity, and specificity of genotypic algorithms in comparison with the original Trofile assay in a selection of non-B HIV-1 samples from the Pfizer A4001064 study.
Materials and Methods
Ethics statement
This study was approved by the University of British Columbia/Providence Health Care Research Ethics Board (H07-01901). The experiments were conducted with the understanding and written consent of each participant.
Samples selection and V3 sequencing
Our study design is outlined in Fig. 1. Samples were selected nonrandomly from the above-mentioned epidemiology study (Pfizer A4001064) to represent large numbers of both phenotypic DM/X4 and R5 viruses (where possible). Samples were tested using a previously described triplicate population-based V3 sequencing approach 11 with automated chromatogram analysis (N=260). Samples (N=280) were also tested with a 454 “deep” sequencing method using a Roche/454 Genome Sequencer FLX (GS-FLX) by combining the triplicates, as previously described. 12

Study overview. Samples were selected from the Pfizer A4001064 epidemiologcal study (N=736) to represent a large number of R5 and non-R5 viruses from each subtype (subtype A, N=95, subtype C, N=105, subtype D, N=106). In the population sequencing analysis, subjects who did not yield Trofile results (unblinded only in the analysis stage) and those with only one success in the triplicate sequencing who had R5 viruses were excluded (N=260). V3 sequences from all replicates were fed into g2p[coreceptor], PSSM X4/R5, PSSM SI/NSI, and PSSM subtype C. The worst-case-scenario approach was taken: Among the replicates, the one with the score that predicts the highest likelihood of being non-R5 would be chosen to represent the genotypic tropism of the sample. In the “deep” sequencing portion of the study, first-round polymerase chain reaction (PCR) amplicons from the same set of samples and replicates (N=301) underwent “nested” PCR with 454-adapted second-round PCR primers and were put through the 454 “deep” sequencing pipeline. N=280 subjects yielded V3 sequences that successfully passed the g2p[454] alignment and tropism prediction pipeline.
Tropism determination
Population V3 sequences were aligned and analyzed using various bioinformatics algorithms in combination with different cutoffs shown in parentheses: g2p[coreceptor] (5.75% and 10% false-positive rate or fpr), PSSM X4/R5 (–4.25), PSSM SI/NSI (–1.75), and PSSM subtype C (–21.64) developed specifically for subtype C HIV-1. These genotypic tropism prediction cutoffs for population sequencing were optimized in previous studies based on MOTIVATE-1/-2 and A4001029 to best predict clinical outcomes on maraviroc-containing therapies 11,13,14 or were obtained through personal communications with the developer of the PSSM subtype C matrix. “Deep” 454 V3 sequences were aligned using either g2p[454] or an in-house algorithm, Emeline version 2.4. Aligned sequences were scored with either g2p[454] or the in-house g2p Scorer version 1.1. Since 454 “deep” sequencing tropism determination is an independent assay from population sequencing tropism determination, the cutoff for “deep” sequencing results that best predicted the maraviroc clinical outcome for this assay was used. In our previous studies, we reported an optimized g2p[454] cutoff that defined a “non-R5 sample” as “a sample having larger than or equal to 2% non-R5 sequences defined by having a g2p fpr smaller than or equal to 3.5%.” 12,15,16,17 Phenotypic tropism was determined by the original Trofile assay; DM and X4 categories were combined as “non-R5.” It is important to note that neither ESTA data nor post-MVC virological outcome was available to be included in the study.
Results
Genotypic tropism prediction of Trofile results had similar performances in subtype B and collectively analyzed non-B samples
All non-B samples (subtypes A, C, and D) were initially analyzed collectively. Population sequencing predictions by g2p[coreceptor] 5.75% fpr of these non-B samples (N=260) showed 84.2% concordance, 78.5% sensitivity, and 88.2% specificity against the original Trofile assay predictions (Table 1, population sequencing non-B column and Fig. 2b). 454 “deep” sequencing by g2p[454] of these non-B samples (N=280) with cutoff 3.5% fpr and ≤2% non-R5 prevalence showed 86.8% concordance, 92.0% sensitivity, and 82.6% specificity against Trofile predictions (Table 1, 454 “deep” sequencing non-B column). The above results were compared to subtype B results from the MOTIVATE studies.
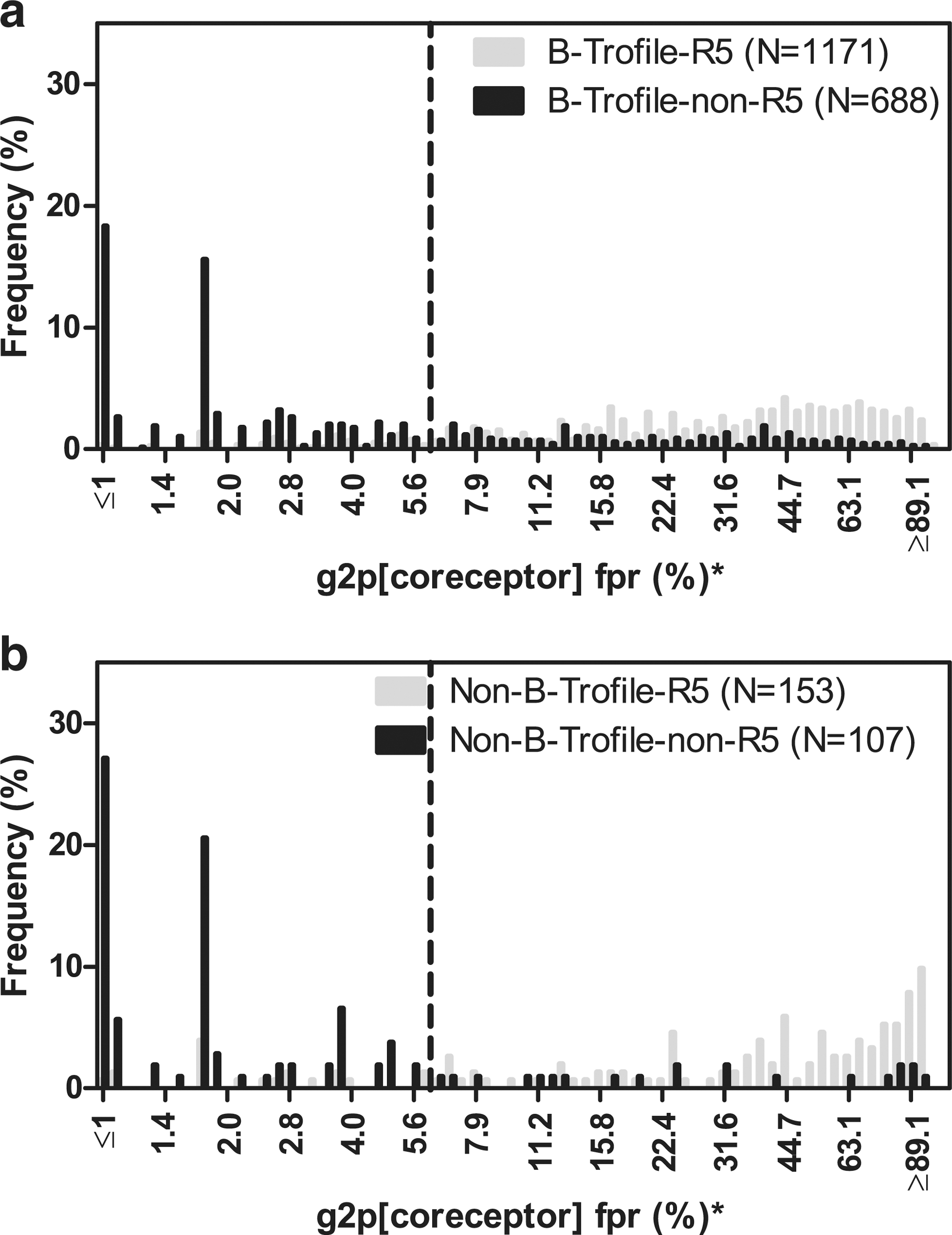
Histograms of population V3 sequencing g2p fpr (%) distribution, stratified by Trofile-determined tropisms (gray, R5; black, non-R5).
fpr, false-positive rate.
In a previous report 11 we demonstrated that population sequencing-based tropism prediction algorithm g2p[coreceptor] (cutpoint 5% fpr) predicted the original Trofile assay results in MOTIVATE-1 and −2 with a concordance of 88.9%, sensitivity for non-R5 prediction of 67.4%, and specificity for non-R5 prediction of 92.6% (N=1,164, 94% subtype B). Despite the relatively poor sensitivity, population-based V3 sequencing was consistent in predicting clinical outcome. In the current study, we reanalyzed the MOTIVATE data by including all screening samples (regardless of entry into the trials) and excluding all non-B samples. This increased our subtype B sample size to N=1,859. The cutpoint for the g2p[coreceptor] was later optimized to 5.75% fpr, based on virologic outcome in the MOTIVATE trials. 13 Using this cutpoint, we observed similar concordance of 81.3%, sensitivity of 65.6%, and specificity of 90.5% in predicting Trofile results (Table 1, population sequencing subtype B column and Fig. 2a). With a less stringent cutpoint at 10% fpr for non-R5 prediction, we observed 78.6% concordance, 74.0% sensitivity, and 81.4% specificity for predicting original Trofile results.
Tropism predictions by 454 “deep” sequencing for the subtype B portion of the MOTIVATE dataset (N=1,823) were also compared to the original Trofile assay (cutpoint 3.5% fpr and 2% non-R5 prevalence), and we observed 82.3% concordance, 80.0% sensitivity, and 83.6% specificity (Table 1, 454 “deep” sequencing subtype B column). With a less stringent cutpoint at 3.5% fpr and ≥5% non-R5 prevalence, we observed 82.0% concordance, 73.5% sensitivity, and 86.9% specificity for subtype B samples, and 84.6% concordance, 83.2% sensitivity, and 85.8% specificity for non-B samples. In summary, the overall concordance and sensitivity of collectively analyzed non-B samples against Trofile predictions were slightly better than subtype B samples.
Population sequencing subanalysis by HIV-1 subtype
g2p[coreceptor]
When using g2p[coreceptor] to predict tropism from population sequences (Table 2, population sequencing section), there was a large decrease in sensitivity by both cutpoints 5.75% and 10% fpr in subtype A compared to subtype B (44.4% vs. 65.6% and 55.6% vs. 74.0%, respectively), but had increased specificity and concordance (Table 2, note b). A detailed g2p[coreceptor] score distribution of subtype A (N=78) samples, stratified by their Trofile results, is provided in Supplementary Fig. S1 (Supplementary Data are available online at
Subtype B data are obtained from MOTIVATE/1029 studies.
Represents a decrease from subtype B.
The lower sensitivity of population-based sequencing may partly be explained by a lower percentage of non-R5 variants within subtype A samples. Indeed, among samples that were deemed DM or X4 by Trofile, 454 “deep” sequencing revealed that the percentage of non-R5 in subtype A was significantly lower than subtype C and D (Kruskal–Wallis test, p=0.0125) (Supplementary Fig. S2). It should be noted that this conclusion is limited by the relatively fewer Trofile-predicted DM/X4 subtype A samples available in this study (population sequencing N=9, 454 “deep” sequencing N=12).
In contrast, subtype C genotypic prediction outperformed B in concordance, sensitivity, and specificity relative to Trofile (Table 2 and Supplementary Fig. S1), while subtype D had a lower specificity (Table 2, note b). Compared to subtypes A, B, and C, the subtype D samples that were tested had relatively more samples that were R5 by Trofile but had low fpr scores (i.e., were more non-R5-like) in g2p[coreceptor] (Supplementary Fig. S1).
PSSM
PSSM is an alternative algorithm to g2p[coreceptor] for genotypic tropism prediction. Non-B V3 loop sequences in this study were scored using three versions of the PSSM algorithms: PSSM X4/R5, PSSM SI/NSI, and PSSM subtype C matrices. The distributions of scores are shown in Supplementary Fig. S1. Vertical dotted lines represent optimized cutpoints based on MOTIVATE-1 and −2 trials clinical outcome data (unpublished results). In general, PSSM X4/R5 and SI/NSI matrices show the same trend in score distributions as g2p[coreceptor]. These two PSSM matrices could not clearly differentiate R5 and non-R5 viruses in subtype A and D (Supplementary Fig. S1). Subtype C had a distribution similar to subtype B. Finally, despite the fact that the PSSM subtype C matrix 9 was specifically designed to handle subtype C sequences, in our study it had score distributions similar to the other two matrices but offset toward a lower score range. The optimized cutpoint obtained from personal communication from the developer of PSSM subtype C did distinguish R5 and non-R5 better in subtype C samples than in other subtypes (Supplementary Fig. S1).
454 “deep” sequencing subanalysis by HIV-1 subtypes
Strikingly, 454 “deep” sequencing in conjunction with g2p[454] improved sensitivity for subtype A from the population sequencing results from 44.4% to 83.3% (Table 2). Also of note are the high concordance, sensitivity, and specificity (≥95%) relative to Trofile in subtype C samples. Interestingly, 454 “deep” sequencing did not improve concordance or specificity of subtype D samples. A summary of the distributions of 454-predicted percentage of non-R5 variants, stratified by subtype, is shown in Fig. 3.

Scatter plot of the distribution of % non-R5 per sample as determined by 454 “deep” sequencing and g2p[454], grouped by Trofile-predicted tropism and subtypes. Medians and interquartile ranges are shown in black.
Discussion
This study provided previously unavailable data on bioinformatic algorithms for predicting viral tropism in non-B HIV-1 subtypes. Genotypic assays had similar overall concordance with the original Trofile assay and with each other in these non-B subtypes as they did for subtype B, but subdividing into the specific non-B subtypes could reveal different performances.
This study was limited by the fact that (1) comparisons were based on the original Trofile assay instead of the newer, more sensitive ESTA, (2) no clinical outcome information was available for patients infected with non-B viruses receiving CCR5-antagonist-based therapy, and (3) the Trofile-predicted R5 and DM/X4 could not be evenly represented in this study, nor were the samples from each subtype evenly distributed (Table 2).
In this study, we observed that sensitivities and specificities relative to the original Trofile differ among the HIV-1 subtypes. Using g2p[coreceptor], specificity was lowest with subtype D, while sensitivity was lowest for subtype A. The PSSM algorithms also distinguished non-R5 from R5 variants, depending on the cutpoints used. These observations suggest that if the goal is to predict the results of the original Trofile, population sequencing algorithms cutpoints can be manipulated according to the histograms presented here to further optimize sensitivity and specificity for each subtype. These results imply that the clinical cutpoints optimized for subtype B may not necessarily work well with the other non-B subtypes. Ideally, however, concordance with post-MVC virological outcome in patients infected with non-B HIV-1 subtypes, instead of Trofile results, will provide the most clinically relevant evidence for the applicability of these genotypic assays in clinical settings.
We have also found that 454 “deep” sequencing reveals that non-R5 virus appears to be relatively rare in subtype A individuals, and when detected appeared to be present at a low prevalence within individuals. This can potentially become a limitation in clinical detection of non-R5 viruses within subtype A samples and should be further addressed in future studies. However, the small sample size, especially of non-R5 subtype A, is a limitation of this conclusion.
We also observed low specificity in subtype D relative to subtype B in both population and 454 “deep” sequencing. Since 454 “deep” sequencing is more sensitive in the detection of minority species than population sequencing, this observation suggests that the low specificity may be an artifact of the matrices used in this study, rather than the sensitivity of the assay. This is an especially important concern for subtype D infections as it has the highest prevalence of non-R5 viruses and has been observed to harbor phenotypically dual-tropic (non-R5) viruses that have identical V3 loop sequences with cocirculating phenotypically R5 virus, implying tropism determinants outside of the V3 loop. 18
Other studies have also investigated the performance of these genotypic algorithms in non-B samples. For subtype C viruses, it has been shown that both PSSM and g2p[coreceptors] developed for subtype B viruses had sensitivities and specificities comparable to subtype B predictions (N=52). 19 In contrast, subtype D viruses suffer low specificities (N=32) and a new algorithm has been built specific to this subtype. 20 In general, our study yielded similar results but had a larger sample size and included 454 “deep” sequencing comparisons.
In conclusion, our current study showed that although the general concordance with Trofile among non-B samples is comparable to the clinically optimized subtype B results, genotypic tropism predictions in non-B HIV-1, especially in subtype A and D samples, must be interpreted with some caution. Alternative cutoffs that are different from those optimized for subtype B should be considered for each of the non-B subtypes. If possible, future studies should include post-MVC virological outcome data from patients infected with non-B viruses in combination with a larger sample size of these genotypic predictions in order to provide clinically relevant evidence in guiding clinical decisions.
Footnotes
Acknowledgments
We would like to thank all patients enrolled in this study. We would also like to thank Mr. Conon Woods and Mr. Chanson Brumme for bioinformatics support. Contributions of the authors: G.Q.L.: manuscript preparation. G.Q.L., P.R.H., A.F.Y.P., H.V., D.C., J.H.: study design, result analysis, final review. W.D.: HIV-1 genotyping, final review. J.H., J.D., A.R., D.C., H.V., S.P.: subject recruitment, samples preparation, final review. This work was funded by Canadian Institutes of Health Research (CIHR), Pfizer, and ViiV Healthcare. A.F.Y.P. is supported by a Canadian Institutes of Health Research (CIHR) Operating Grant (201009HOP-235256).
Presented in part at the 20th International Workshop on HIV and Hepatitis Virus Drug Resistance and Curative Strategies, June 7–10, 2011, Los Cabos, Mexico (Abstract 78).
Author Disclosure Statement
P.R.H. has received consulting fees from ViiV/Pfizer and Quest and holds stock options in Merck.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.