
Abstract
Memory CD4+ T lymphocytes in peripheral blood that express integrins α4ß7 preferentially recirculate through gut-associated lymphoid tissue (GALT), a proposed site of significant HIV-1 replication. Tregs and activated CD4+ T cells in GALT could also be particularly susceptible to infection. We therefore hypothesized that infection of these subsets of memory CD4+ T cells may contribute disproportionately to the HIV-1 reservoir. A cross-sectional study of CD4+ T cell subsets of memory CD45RO+ cells in peripheral blood mononuclear cells (PBMCs) was conducted using leukapheresis from eight subjects with untreated chronic HIV-1 infection. Real-time polymerase chain reaction (PCR) was used to quantify total and integrated HIV-1 DNA levels from memory CD4+ T cells sorted into integrin β7+ vs. β7−, CD25+CD127low Treg vs. CD127high, and activated CD38+ vs. CD38−. More than 80% of total HIV-1 DNA was found to reside in the integrin β7-negative non-gut-homing subset of CD45RO+ memory CD4+ T cells. Less than 10% was found in highly purified Tregs or CD38+ activated memory cells. Similarly, integrated HIV-1 DNA copies were found to be more abundant in resting non-gut-homing memory CD4+ T cells (76%) than in their activated counterparts (23%). Our investigations showed that the majority of both total and integrated HIV-1 DNA was found within non-gut-homing resting CD4+ T cells.
Introduction
T
Much evidence indicates that gut-associated lymphoid tissue (GALT) plays a major role in the pathogenesis of progressive HIV-1 infection. GALT is believed to contain a large majority of the CD4+ T cells in the body, 12 which are mostly CCR5+13 and in an activated state, 14 making them highly susceptible to infection and depletion. 15 Following this early depletion, chronic HIV-1 infection is believed to result in increased microbial translocation and increased activation, heightening susceptibility of more CD4+ T cells to infection and continuing depletion 15 ; however, there are conflicting data regarding this theory. 16 Furthermore, a recent report has suggested continuing replication of HIV-1 in GALT despite suppressive ART, 17 and supporting this observation, treatment intensification has been reported to reduce viral replication in GALT tissue biopsies. 18 It is therefore plausible to expect that a large number of memory CD4+ T cells containing HIV-1 DNA are present in cells trafficking through the GALT.
Resting memory CD4+ T cells have specialized migratory capacities determined by their expressed integrins. 19 Those generated in GALT in the presence of metabolites of vitamin A express the integrin ß7, 20 which is expressed in conjunction with α4. 19,21 –23 These cells recirculate through mucosal surfaces, such as the genitourinary tract and respiratory tree, and traffic through GALT from peripheral blood, 20,24,25 via specific binding of integrin α4ß7 to MAdCAM-1, which is expressed on specialized endothelial cells in GALT 20,26 and other mucosal surfaces involved in inflammation. 26,27 Memory CD4+ T cells in peripheral blood can be subdivided into two main subsets based on integrin expression, gut-homing α4ß7+ and non-gut-homing α4ß1+ cells. The latter cells cannot access GALT since they cannot bind MAdCAM-1. 20
T regulatory CD4+ cells (Tregs) reduce the effects of proinflammatory stimulus created by gut microbials. 28,29 Hence microbial translocation occurring during chronic HIV-1 infection would likely increase Treg cells and possibly allow for their infection by HIV-1. Increases in Foxp3+ Tregs in mucosal tissue in chronic HIV-1 infection have been demonstrated. 30,31 Tregs, originally defined as CD25high CD4+ T cells, have also been reported to be susceptible to HIV-1 infection in vitro. 32 Currently there is little agreement in terms of their expansion or depletion during HIV-1 infection in vivo. 33 –41 With increased activation and infection of the remaining CD4+ T cells following the depletion of CD4+ T cells in the GALT, it is possible that Tregs are affected. 15 However, their survival during HIV-1 disease progression and their presence in mucosal tissues 42 suggest that they traffic through the GALT and hence may play a role as a viral reservoir.
Integrin ß7+ CD4+ T cells were sorted from peripheral blood mononuclear cells (PBMCs) of untreated, chronically HIV-infected subjects to determine whether they preferentially contained HIV-1 DNA. We have also used the improved cell surface definition of Tregs, CD25+CD127low, for purification of human Tregs from PBMCs by cell sorting, 43 to allow us to better clarify the extent to which Tregs are infected in vivo with HIV-1 DNA. Finally, by sorting activated CD38+ memory CD4+ T cells, we have assessed whether there was preferential infection of these cells in chronic untreated HIV-1 infection.
Materials and Methods
Patients
Eight treatment-naive subjects with documented chronic HIV-1 infection (CHI) and relatively high CD4+ T cell counts in peripheral blood were included in this study (Table 1). Leukapheresis packs were collected between 1992 and 1994, and cells were cryopreserved as part of a study of adoptive immunotherapy. 44 However, these samples preceded routine plasma viral load testing, and no results are available for HIV RNA copy numbers for the times of collection.
IQR, interquartile range.
In addition, gut-homing and non-gut-homing memory CD4+ T cell subsets were isolated from two PHI patients, both with HIV-1 viral load levels >750,000 copies/ml, as previously described, 45 and two patients on cART for greater than 6 months, with plasma viral loads <50 copies/ml. 46 Investigation of sorted CD4+ T cells was restricted to gut-homing and non-gut-homing memory CD4+ T cell subsets for these two groups of patients.
Purification of memory CD4+ T cell subsets
Following thawing of leukapheresis packs, cell viability was routinely >80%. After enrichment with Dynabeads Untouched Human CD4 T Cell kit (Invitrogen, Oslo, Norway), 1–2×108 enriched CD4+ T cells were stained using the following three panels of monoclonal antibody cocktails into 14 subsets: CD3-PerCP-Cy5.5, CD4+-PE-Cy7 (BD Biosciences, San Jose, CA), and CD45RO-ECD (Beckman Coulter, Hialeah, FL) were used in all three panels to define CD4+ T cells and to mark memory phenotype.
In panel 1, CD27-FITC was added and combined with CD45RO-ECD to gate naive cells. Integrin β7-PE (BD Biosciences) was added to collect gut-homing (β7+) and non-gut-homing (β7−) memory cells. CD25-APC (BD Biosciences) and CD127-eFluor450 (eBioscience) were combined for sorting naive and memory regulatory T cells (CD25+CD127low) and CD127+ cells.
In panel 2, CCR7-PE (purified CCR7 IgM, BD Biosciences plus anti-IgM-PE (antimouse μ chain-specific, Jackson ImmunoResearch, West Grove, PA) and CD27-FITC (BD Biosciences) were used to define central memory (CD45RO+CCR7+ or CD45RO+CD27+), effector memory (CD45RO+CCR7−CD27− or CD45RO+CD27−), transitional memory (CD45RO+CCR7−CD27+), and naive and terminally differentiated (CD45RO−CD27−) CD4 cells.
In panel 3, CD38-APC (BD Biosciences) was used to collect two memory populations (CD45RO+CD38+ and CD45RO+CD38−). Cell–antibody mixtures were incubated at room temperature for 15 min then washed with PBA (DPBS with 10% bovine serum albumin and 2% azide) and fixed with 0.5% PFA. Cell suspensions were filtered through a 70-μm cell strainer and adjusted to a density of 20×106/ml ready for sorting. CD3+CD4+CD45RO+ memory cells were sorted into subsets using a FACSAria (Becton Dickinson) with >96% purity. A minimum of 200,000 purified cells was obtained for each subset.
Total and integrated HIV-1 DNA real-time quantification
Total DNA was extracted from sorted CD4+ subsets using the Qiagen DNeasy Blood and Tissue kit (Hilden, Germany) and total HIV-1 DNA was quantified by a real-time polymerase chain reaction (PCR) assay specific for HIV-gag 47 with a lower limit of detection (LLOD) of 10 copies and a CV of 11%. 10 A standard dilution curve of pNL4-3 plasmid from 107 through to 101 copies was used as an internal control. Briefly, 800 nM of the sense primer SK145 5′-AGTGGGGGGACATCAAGCAGCCATGCAAAT-3′ and the antisense primer SKCC1B 5′-TACTAGTAGTTCCTGCTATGTCACTTCC-3′ was used in conjunction with 200 nM of the dual-labeled fluorogenic TaqMan locked nucleic acid probe SKLNA2-3 5′-6-FAM AT[C]A[A]T[G]AGGAA[G]CT[G]C-BHQ-1-3′. For a 25 μl reaction, 12.5 μl of iQ Supermix (Bio-Rad Laboratories, Hercules, CA) was used with 5 μl of DNA template. PCR conditions consisted of one cycle of 95°C for 3 min followed by 40 cycles of 95°C for 15 s and 60°C for 1 min.
Integrated HIV-1 DNA was quantified using real-time PCR with an LLOD of eight copies and a CV of 26% as previously described. 10 In brief, an ACH2 cell line was used for the standard curve. For the first round 300 nM of Alu 1 and 2 primers was used in conjunction with 100 nM of the lambda heel primer L-M667 on an Eppendorf thermocycler (Hamburg, Germany). PCR conditions were one cycle of 94°C for 7 min, 12 cycles of 94°C for 30 s, 60°C for 30 s, and 72°C for 3 min and one cycle of 72°C for 7 min. In the second round 300 nM of primers Lambda T and AA55M were used with 200 nM of a dual-labeled fluorogenic probe, LTR FL, on a Bio-Rad iQ-5 real-time PCR machine. PCR conditions consisted of one cycle of 5 min at 95°C, followed by 45 cycles of 95°C for 15 s and 60°C for 1 min. Samples were run in quadruplicate for the first round PCR and performed in duplicate for the second round PCR.
Total and integrated HIV-1 DNA was compared and normalized with genomic DNA, determined by β-actin detection using the Applied Biosystems (Carlsbad, CA) TaqMan β-actin detection reagents with a FAM-labeled probe. For the β-actin assay, 12.5 μl of iQ Supermix was used in a 25 μl reaction with 180 nM of sense and antisense primers and 120 nM of probe. PCR conditions were as per each assay. The standard curve was constructed from purified human buffy coat DNA and results were expressed as copies/500 ng DNA. Where DNA yield in a subset was inadequate for quantification, these patients were omitted. Therefore the number of patients compared for each subset (n) is given on the corresponding graphs.
Statistical analysis
Percentages of subsets of memory cells within CD4+ T cells were expressed as medians and compared between the HIV-infected subject group by the Mann–Whitney nonparametric test (GraphPad Software, San Diego, CA). Absolute total and integrated HIV-1 DNA copy numbers/500 ng DNA for each subset were expressed as medians and compared. To calculate the relative contribution of each subset to the copy number in memory CD4 T cells, for each individual subject, the absolute HIV-1 DNA copy number median/500 ng DNA for the subset of interest was adjusted to take into account the relative proportion of that subset, as determined by flow cytometric phenotyping. For example, in one subject, ß7+ cells contained 1,166 copies HIV DNA/500 ng DNA. Using the conversion that 500 ng DNA is equivalent to 80,000 cells, then each ß7+ cell contained 0.0146 copies, whereas in the same patient, ß7− cells contained 535/80,000=0.0066875 copies per cell. By flow cytometry, ß7+ cells were 10.5% of CD45RO+ cells, while ß7− cells were 85% of CD45RO+ cells. Therefore for every 100 CD45RO+ CD4+ T cells, ß7+ cells contributed 10.5×0.0146=0.1533 copies, while ß7− cells contributed 85×0.0066875=0.568 copies. This corresponds to ß7+ contributing 0.1533/0.7213=21% of copies and ß7− cells contributing 0.568/0.7213=79% of copies. These calculations were performed for each individual subject, for each pair of subsets of CD45RO+ cells, namely ß7+ vs. ß7−, Treg vs. CD127high, and CD38+ vs. CD38− for each individual. Two-tailed Wilcoxon matched-pairs signed rank tests were used to compare between paired subsets for all individuals (GraphPad Software, San Diego, CA).
Results
Immunophenotyping of subsets of memory CD4+ T cells
The eight CHI subjects included in this study were treatment naïve and infected for a median of 5 years of infection since diagnosis with relatively high CD4+ T cell numbers (Table 1). No HIV-1 plasma viral load data were available for these patients at the time of collection, although their median CD4+ cell count of 622 cells/μl was significantly lower than the median for HIV-1-uninfected controls (1053 cells/μl, p<0.001), clearly indicating early progressive disease.
In initial cell sorting experiments, the rate of infection of the CD45RO− subset was substantially lower (≈10 fold) compared to the CD45RO+ subset, consistent with previous reports. 45,48,49 Therefore, staining panels were rationalized to allow for a better understanding of subsets thought to contribute most substantially to HIV-1 viral reservoirs. As a result, the final panel included CD45RO+β7+ (gut-homing) vs. CD45RO+β7− (non-gut-homing) cells, CD45RO+CD25+CD127low (Tregs) vs. CD45RO+CD127high cells, and CD45RO+CD38+ (activated memory) vs. CD45RO+CD38− (resting memory) cells.
The phenotyping results showed that gut-homing ß7+ cells, Tregs, and activated cells are a minority of CD45RO+ memory CD4+ T cells in peripheral blood. Gut-homing ß7+ cells are outnumbered greater than 5-fold by non-gut-homing ß7− memory CD4+ T cells, Tregs outnumbered over 9-fold by CD127high memory CD4+ T cells, and CD38+ activated cells outnumbered over 11-fold by resting CD38− memory CD4+ T cells, respectively (Table 1).
Gut-homing versus non-gut-homing CD4+ T cells
CD4+ memory T cells that express integrin α4 reciprocally express either β1 or β7 integrin (Fig. 1A). Therefore we identified gut-homing memory CD4+ T cells as CD45RO+ integrin ß7+, and non-gut-homing memory CD4+ T cells as CD45RO+ integrin ß7−(Fig. 1A).
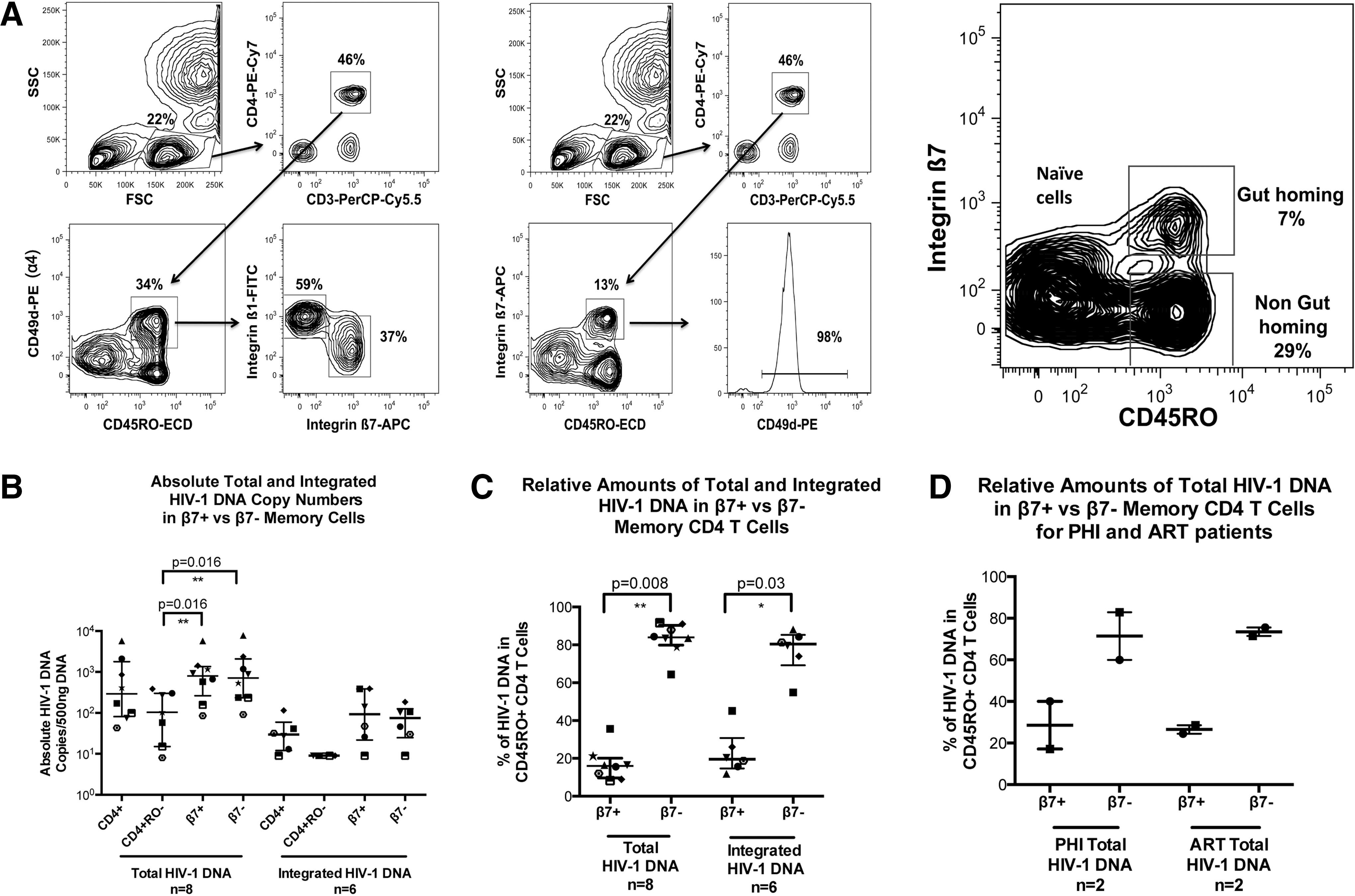
Gut-homing memory CD4+ T cell population in antiretroviral treatment (ART)-suppressed primary HIV-1 infection (PHI) and chronic HIV-1 infection (CHI) patients. Memory gut-homing CD4+ T cells were identified by expression of CD45RO and integrin β7.
Total and integrated HIV-1 DNA were quantified for purified CD4+ T cells, naive CD4+ T cells (CD3+CD4+CD45RO−), gut-homing memory CD4+ T cells (CD3+CD4+CD45RO+β7+), and non-gut-homing memory CD4+ T cells (CD3+CD4+CD45RO+β7−) for CHI patients (Fig. 1B). Absolute copy number medians showed that there was no preferential infection of the gut-homing CD4+ T cells compared to their non-gut-homing counterparts. In contrast, the naive CD4+ T cell population median was lower in both DNA species than either the CD4+ T cell or memory population medians, and was significantly lower upon comparison of total HIV-1 DNA contained within either β7 subset (p=0.016).
However, when cell proportions were taken into account, the gut-homing memory CD4+ T cell subset contributed only 16% of all total HIV-1 DNA copies and 19.5% of all integrated HIV-1 DNA copies in memory CD4+ T cells (Fig. 1C), significantly different from non-gut-homing memory CD4+ T cells (p=0.008 and p=0.03 for total and integrated HIV-1 DNA, respectively). A similar distribution of total HIV-1 DNA was found in both acute untreated PHI patients and the CHI patients on suppressive ART (Fig. 1D).
Tregs versus long-term memory CD4+ T cells
Memory CD4+ T cells were stained and sorted for memory Treg phenotype (CD45RO+CD25+CD127dim) and compared to memory CD127high CD4+ T cells (CD45RO+CD127high), as shown in Fig. 2A. The memory CD127high CD4+ T cells were shown to be 10 times more abundant than the memory Treg CD4+ T cells.
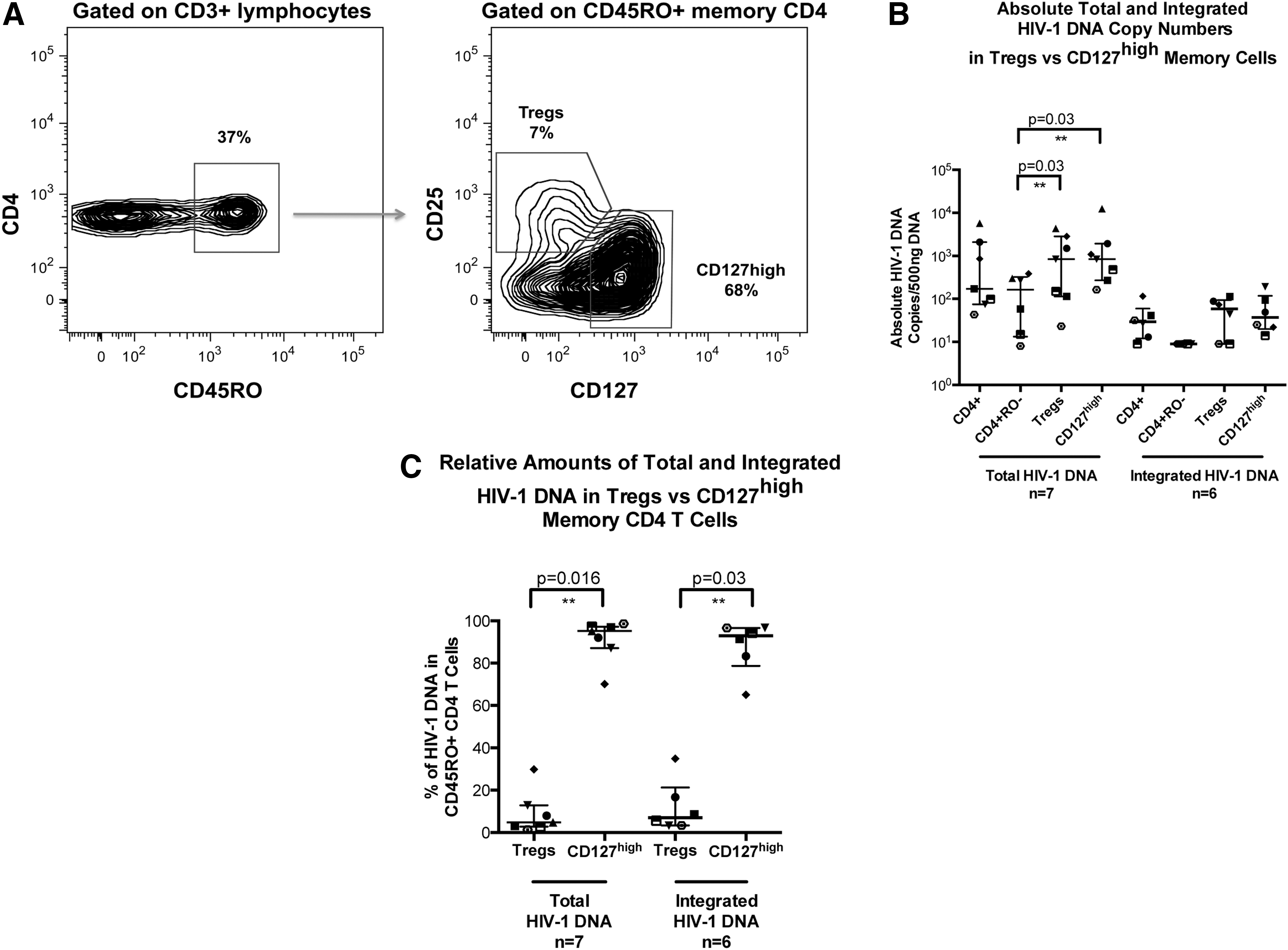
Memory T regulatory and memory CD127high CD4+ T cell subsets in CHI. Memory Treg CD3+CD4+ cells were identified within the CD45RO+ population by high expression of CD25 and dim expression of CD127.
Absolute total and integrated HIV-1 DNA quantification showed a similar distribution of HIV-1 DNA copies/500 ng between the two populations, with no evidence of preferential infection of Tregs (Fig. 2B). However, both populations significantly contained greater levels of total HIV-1 DNA compared to naive CD4+ T cells (p=0.03). Memory Treg CD4+ T cells were shown to contribute a median of 5% of total HIV-1 copies, compared to 95% contributed by CD127high memory CD4+ T cells (p=0.016; Fig. 2C). Similarly, only 7% of integrated HIV-1 DNA copies were contained within memory Tregs, compared to 93% of copies found in the CD127high memory CD4+ T cell population (p=0.03, Fig. 2C).
Activated CD38+ memory CD4+ T cells versus resting CD38− memory CD4+ T cells
Memory CD4+ T cells were stained and sorted based on the activation marker CD38+, as shown for PHI and CHI patients, respectively, in Fig. 3A. During PHI the level of activation is increased (as we previously reported 50 ) but eventually plateaus at around 10% in untreated CHI, similar to donor 2 for this study (Fig. 3A).
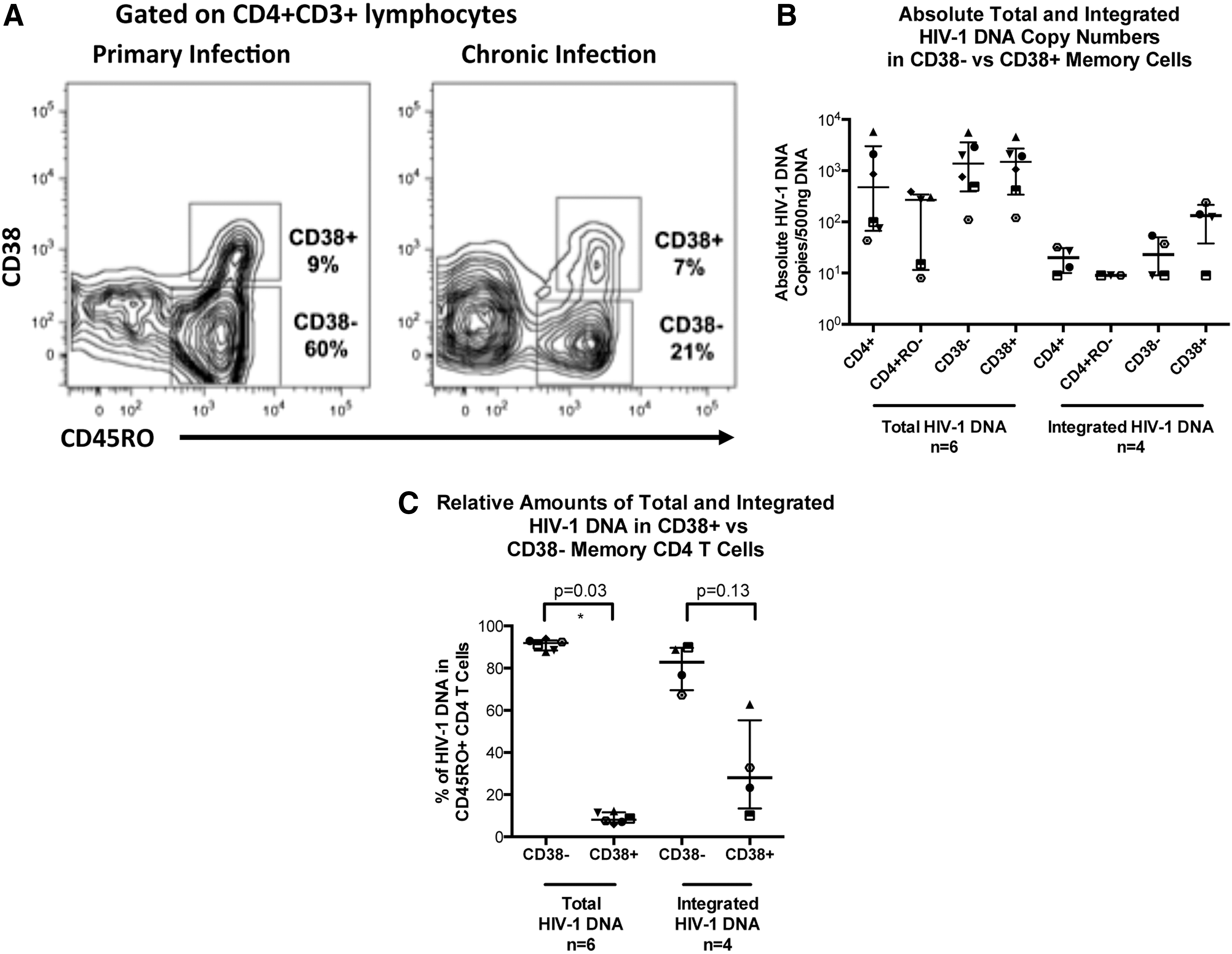
Activated memory cells identified by their expression of CD38.
Quantification of total HIV-1 DNA in activated CD38+ memory CD4+ T cells showed that there was no preferential infection of this subset (Fig. 3B). Taking into account the proportional differences, we found that the majority of total HIV-1 DNA was significantly contained within the resting CD38− population (p=0.03; Fig. 3C). A median of 92% of all CD4+ T cell total HIV-1 DNA was within this population, with a median of only 8% within the activated CD38+ population.
Integrated HIV-1 DNA copies in the activated CD38+ memory CD4+ T cell population were higher than in the resting CD38− memory CD4+ T cells in the four samples quantified. The median absolute copy numbers were 3.5-fold higher in the activated CD38+ memory CD4+ T cells than in the resting CD38− memory CD4+ T cells (Fig. 3B). Even so, upon consideration of cell numbers, the CD38− population was still shown to comprise the majority of integrated HIV-1 DNA, containing a median of 76% (Fig. 3C); however, due to the low sample number these differences in integrated HIV-1 DNA were not shown to be significant (p=0.13).
Discussion
To rationally design therapies that target the HIV-1 reservoir, it is essential to improve our understanding of which cell subsets contain proviral HIV-1 DNA and to further delineate the mechanisms of how this reservoir is generated and maintained. Recently, progress has been made in studies characterizing the infection of central versus effector memory CD4+ T cells in peripheral blood. 49 However, to date, the contribution of circulating gut-homing memory CD4+ T cells in peripheral blood to the HIV-1 reservoir has been studied only in vitro. 25 Similarly, study of infection of resting cells has generally used the lack of expression of HLA-DR, CD25, and CD69 as markers of activation. 51 Infection of activated CD38+ CD4+ T cells in PBMCs in vitro and lymphoid tissue in vivo has been investigated 52 ; however, circulating CD38+ activated memory CD4+ T cells, which are pathognomonic of progressive chronic HIV-1 infection, 53 have not been studied in detail.
Research has focused on HIV-1 replication and CD4+ T cell depletion in GALT as one of the major drivers of HIV-1 pathogenesis and subsequent immunodeficiency. 15,54 Experimental evidence strongly suggests that CD4+ T cells from GALT and other mucosal surfaces mostly recirculate through efferent lymph to the thoracic duct and thence back into the circulation, before reentering GALT via cell surface expression of integrins α4ß7. 20,55 We therefore tested the hypothesis that the HIV-1 reservoir is, to a significant degree, established and maintained in memory CD4+ T cells associated with GALT, particularly in those cells that express CD38 during untreated chronic HIV-1 infection. We expected these memory CD4+ T cells in peripheral blood to contain higher levels of HIV-1 DNA compared to non-gut-homing memory CD4+ T cells. In particular, we expected that the phenotyping of recently activated CD38+ cells as well as Treg cells may further narrow the definition of the cells that contain a large part of the reservoir within the circulating CD4+ T cell pool.
Surprisingly, our results suggest that the HIV-1 reservoir within circulating memory CD4+ T cells is not preferentially generated by recent activation of these cells within GALT, since only a small minority of HIV-1 DNA was contributed by memory CD4+ T cells that were either gut-homing or that were expressing CD38 and therefore recently activated. Our finding that CD38+ and CD38− memory CD4+ T cells had similar per cell levels of HIV-1 DNA is consistent with our previous findings in PHI, 45 and reports in SIV infection, 56,57 but differs from two other studies of PBMCs and one study of lymph node cells where activated cells were reported to contain more HIV-1 DNA. 51,52,58 In those latter studies, activated CD4+ T cells were compared to nonactivated CD4+ T cells, which presumably contained substantial proportions of naive CD4+ T cells, which contain approximately 10-fold less HIV-1 DNA than memory CD4+ T cells. 45,59 One study defined HLA-DR−CD25−CD69− as resting cells, 51 but we have found that this subset contains CD38+ cells, which were also Ki-67+, consistent with very recent activation and proliferation (data not shown), and consistent with the fact that CD38 elevation on its own represents an independent marker for progressive disease. 53
One possible alternative explanation for our findings is that infection of α4ß7+ cells results in loss of cell surface expression of these integrins, leading to an apparent α4ß7− phenotype. However, we were able to confirm that the reciprocal α4ß1+ subset was clearly infected (data not shown), and since these cells were unlikely to have previously been α4ß7+ a possible loss of α4ß7 expression appears less likely. It would also be conceivable that a significant fraction of HIV-1 DNA containing CD4+ T cells is selectively retained in GALT. However, compelling data from other studies indicate that the initial rise in CD4+ T cell counts after initiation of ART is, to a significant degree, due to the release of memory CD4+ T cells retained in sites such as peripheral lymph nodes, 60 and it seems reasonable to expect that the same mechanism applies to all secondary lymphoid tissues.
In a recent analysis of patients who have initiated therapy with raltegravir-containing ART during either primary or chronic HIV-1 infection we did not observe significant changes in the proportions of circulating α4ß7+ memory CD4+ T cells after commencing therapy, indicating no preferential retention of these cells subset. 10 It has also been reported that HIV-1 uses a tripeptide motif in the V2 region of Env to engage α4ß7 on the surface of CCR5+ CD4+ T cells, increasing entry into target cells in vitro, 61 particularly during PHI. However, these data have been recently contested with tropism shown to be restricted to a limited number of quasispecies. 62 Our findings suggest that this mechanism does not significantly contribute to the relative amounts of HIV-1 DNA found in peripheral blood subsets.
Our direct comparison of gut-homing to non-gut-homing memory CD4+ T cells showed no significant difference between these two subsets on a per-cell basis similar to one other study in vitro. 25 Other studies, however, have indicated relatively high levels of HIV-1 DNA in CD4+ T cells from GALT biopsies compared to PBMCs, 14,17,63 presumably due to the presence of activated CCR5+ CD4+ T cells as target cells in GALT biopsies. 14 However, earlier studies of CD4+ T cells isolated from peripheral lymph nodes also showed high levels of activated CD4+ T cells 64 as well as high levels of HIV-1 DNA, 65 relative to PBMCs, similar in magnitude to the GALT versus PBMC comparisons.
Our study confirms and extends our previous finding that most of the HIV-1 DNA in PBMCs is found in CD4+ T cells that are CD127high, 45 which is consistent with the recent suggestion that interleukin-7 (IL-7) signaling is a major mechanism of maintenance of the reservoir. 49 In contrast, in Tregs, which are CD127low, we confirmed infection with HIV-1, but not preferential infection, since this subset did not contain increased amounts of HIV-1 DNA compared to CD127high cells; however, both subsets contained approximately a log greater number of HIV-1 DNA copies/500 ng DNA when compared to CD45RO− cells. We were able to detect HIV-1 DNA in highly purified CD45RO+ memory Tregs, but not in the CD45RA+ naive Tregs subset 36 (data not shown). This is consistent with low, but detectable expression of CCR5 on a small subset of memory Tregs (data not shown). It is still debatable whether Tregs are relatively expanded or depleted in CHI, 66 but our current data would suggest that there is not preferential depletion due to infection.
Taken together the results from our study suggest that the exact contribution of the GALT to the HIV-1 reservoir will require further exploration. It is likely that the ability of these subsets to produce infectious HIV-1 upon reactivation will likely differ, which is beyond the scope of this study. However, it was recently shown that despite the role of GALT as an HIV-1 viral reservoir, it was not the major source of rebounding virus 67 ; nevertheless, further investigations are required, especially if reactivation of the viral reservoir is to be incorporated into therapeutic interventions. We did not find preferential infection of, or higher levels of, HIV-1 DNA in peripheral gut-homing memory CD4+ T cells. Recently, Ganusov and De Boer have suggested that GALT may contain only a minority of all the memory CD4+ T cells in the body at any one time. 68 Therefore, if the infection rate in memory CD4+ T cells in GALT is comparable to the infection rate in other lymphoid tissues, as supported by our results, it would be plausible that only a small fraction of the HIV-1 reservoir exists in GALT at any one time. To further clarify this point, direct quantification of HIV-1 DNA in these CD4+ T cell subsets from gut mucosa would be highly desirable; however, in our experience, the accurate quantification of total HIV-1 DNA in gut mucosa is problematic, as the minimum cell numbers required for quantification from sorted cells are difficult to attain from standard pinch biopsies (unpublished data). Other studies nevertheless have shown a decreased number of CD4+ T cells compared to peripheral blood but a higher proviral burden. 17,69,70 Others again have also found correlations between HIV DNA burden in mucosal tissues and PBMCs, in those on HAART, 71 although this is also a point of divergence. 63
There are several limitations associated with this study, which should be noted. First the lack of HIV-1 plasma viral load (pVL) did not allow for correlations between the level of viral DNA within a subset and HIV-1 pVL to be made. While memory stem cells are now thought to be an important contributor to the HIV-1 viral reservoir, 72 these cells were described only during the conduct of the study outlined above and hence for reasons of consistency, the panel remained unchanged and does not include these cells. A further restraint was the fact these patients were untreated and were not subsequently followed over the course of infection; we were therefore unable to confirm if the differences seen between the HIV-1 DNA levels within subsets were maintained during therapy. The HIV-1 DNA real-time PCRs used within this study are capable only of quantifying HIV-1 DNA and cannot distinguish between HIV-1 DNA that is replication competent or incompetent; however, it is also clear that outgrowth assays may not detect all replication-competent virus. 73 This study also did not explore the quantification of HIV-1 DNA within memory CD4+ T cells in patient tissue, due to the lack of access of such material determined by the period of sampling.
Other sites of infection may indeed be secondary lymphoid tissues, which contain large amounts of virus attached to follicular dendritic cells, as well as many productively infected cells, as shown by PCR and in situ hybridization. 74 In particular, the spleen can contain more than 107 infected cells at any one time, 75 with estimates of HIV-1 DNA detected in up to 10% of CD4+ T cells in lymphoid tissue. 74 Together with increased expression of proinflammatory cytokines in HIV-infected lymph nodes 76,77 these tissues may contribute to the maintenance of the pool of latently infected resting CD4+ memory T cells, which recirculate into the periphery. To this end, exposure to IL-7 78 may promote infection of resting CCR5+CD127+ CD4+ memory T cells, or extravasating lymphocytes may be infected following stimulation with chemokines as they enter lymph nodes. 6 In considering possible interventions to target the HIV-1 reservoir, it will be important to analyze all secondary lymphoid tissues.
Footnotes
Acknowledgments
The authors would like to thank the Australian Immunovirology Research Network for provision of leukopheresis packs. The Kirby Institute is supported by funding from the Australian Government Department of Health and Ageing. This work has been supported by NHMRC Project Grant 510325 (J.Z., J.M., and K.S.) and NHMRC Program Grant 510488 (D.A.C., A.D.K., J.Z., N.S., and J.M.).
Author Disclosure Statement
No competing financial interests exist.