
Abstract
In recent years, the increase of migration from countries where human immunodeficiency virus type 2 (HIV-2) is endemic to industrialized countries has facilitated the spread of the virus in individuals previously unexposed to this threat. In this report, we performed a phylogenetic analysis on pol and env sequences of HIV-2 strains identified in foreigners and native citizens to trace the origin of infection. All but one of the 17 pol gene sequences were classified as group A. HIV-2 strains were aggregated in several clusters depending by the country of origin and/or infection. One patient (1AA) was classified as being infected with a recombinant between HIV-2 group A and HIV-2 group B, because the pol gene sequence was clearly in the group A, but an env V3 region sequence from this patient was more similar to group B viruses. Therefore, it is urgent to strengthen the surveillance and use adequate molecular virological tools to diagnose and monitor HIV-2 infection.
Introduction
T
In recent years, Italy, as did other industrialized European countries, welcomed immigrants from countries in Western Africa. Many of these immigrants are not aware of being infected by HIV-2 or do not follow preventive measures to avoid transmission. Thus the risk of acquiring HIV-2 infection is increasing. Hence, the need for improving surveillance through blood screening. Currently in Italy we do not have updated estimates of HIV-2 infection.
When screening for HIV-1/2 antibodies it is recommended that the most advanced diagnostic tests be used. The cross-reactivity between the anti-HIV-2 antibodies and the envelope glycoprotein of HIV-1 could cause a misclassification of HIV-2 patients as being infected with HIV-1. To reduce such mistakes, the HIV-2-specific proteins sgp105 and gp36 have been added to the strip used for Western blot analysis (INNO-LIA HIV I/II Score, Innogenetics, Ghent, Belgium). Urgent also is the need for standardized real-time polymerase chain reaction (PCR) assays able to detect, and quantify accurately, the circulating HIV-2 strains. A correct and timely HIV-2 diagnosis is crucial to ensure appropriate treatment and avoid virus transmission.
In this article we describe 12 cases of HIV-2-infected immigrant patients and analyze the phylogenetic classification of the viruses.
Materials and Methods
Patients
The study included 12 HIV-2-infected patients (nine treatment experienced and three naive). Of these patients, six were from Africa (50%), one from Portugal (9%), one from Cape Verde (9%), and four from India (36.5%). All the patients attended the Infectious Diseases ward in Rome, University of Rome “La Sapienza,” “ Lazzaro Spallanzani” Hospital, Rome, and University Hospital of Rome “Tor Vergata” Italy.
HIV-2 RNA quantification
Plasma HIV-2 RNA levels were measured using an HIV-2 Real Time RT-PCR Kit (Shanghai ZJ Bio-tech Co., Ltd, Shanghai, China). The assay has a lower limit of detection of 5,000 IU/ml. Plasma samples were extracted using the QIAamp Viral RNA Mini kit (Qiagen, Hilden, Germany) and 5 μl was added to the PCR mix according to the manufacturer's instructions.
HIV-2 sequencing
Genotype analyses were performed on plasma and peripheral blood mononuclear cells (PBMCs) using self-made genotypic assay methods developed and optimized in our laboratory. Briefly, the RNA was extracted from plasma samples using the QIAamp Viral RNA mini kit (Qiagen, Hilden, Germany), reverse transcribed, and PCR amplified with SuperScript One-Step RT-PCR for Long Templates (Invitrogen, Milan, Italy) employing specific primers. PCR products were sequenced in sense and antisense orientations using different overlapping sequence-specific primers, a BigDye terminator v. 3.1 cycle sequencing kit (Applied Biosystems, Monza, Italy), and an automated sequencer (ABI-3130). The sequences were analyzed using SeqScape v.2.6 software (Applied Biosystems, Monza, Italy). The quality endpoint for each individual was ensured by coverage of the entire sequences by at least two sequence segments.
DNA extraction from PBMCs was performed using the QIAamp DNA mini kit (Qiagen, Hilden, Germany) according to the manufacturer's instructions. The extracted DNA was amplified using the same primers, PCR conditions, and sequencing methods applied to the plasma samples except for the initial reverse transcription step that was omitted.
Pol gene sequencing
The PCR primers used are listed in Supplementary Table S1 (Supplementary Data are available online at
ENV (V3) gene sequencing
The V3 region was amplified using primers listed in Supplementary Table S1. The conditions for the reaction were as follows: one cycle at 50°C for 30 min; one cycle at 94°C for 2 min; 35 cycles at 95°C for 30 s, 55°C for 30 s, and 72°C for 50 s; and a final cycle at 72°C for 10 min. To increase the yield of the reaction, 5 μl of the amplified product was reamplified with the same primers as the first round of amplification (CFV3S3 and CFV3AS2). The amplified products were sequenced in sense and antisense orientations by using three different overlapping sequence-specific primers. The sequencing primers used are listed in Supplementary Table S1. The reaction conditions were as follows: one cycle at 96°C for 5 min; 25 cycles at 96°C for 30 s, 53°C for 10 s, and 60°C for 4 min.
Phylogenetic analysis
Two different datasets were built. The first dataset included 17 HIV-2 pol gene (PR-RT; 1,278 bp) sequences together with 34 HIV-2 and SIV-SMM reference sequences downloaded from the Los Alamos HIV database (
All the sequences were aligned using MAFFT 9 and were then manually edited with the BioEdit program. 10 For all datasets, the evolutionary model was chosen based on the best-fitting nucleotide substitution model in accordance with the results of the hierarchical likelihood ratio test (HLRT) described by Swofford and Sullivan. 11 Maximum likelihood phylogenetic trees, for the first and second dataset, were generated using the GTR+I+G model of nucleotide substitution, with Phyml v 3.0. 12 The statistical robustness and reliability of the branching order within the phylogenetic trees were confirmed with the bootstrap analysis. The ML trees were rooted by midpoint. The Bayesian phylogenetic tree was reconstructed by means of MrBayes 13 using a general-time-reversible model of nucleotide substitution, a proportion of invariant sites, and gamma-distributed rates among sites (GTR+I+G). A Markov Chain Monte Carlo search was made for 10×106 generations using tree sampling every 100th generation and a burn-in fraction of 25%. Statistical support for specific clades was obtained by calculating the posterior probability of each monophyletic clade, and a posterior consensus tree was generated after a 25% burn-in. The posterior probability was used as a statistical support for each cluster.
Results
Phylogenetic analysis
Maximum likelihood and Bayesian analysis used to determine the HIV-2 groups gave the same results; moreover, the topology of the trees was the same (data not shown). The Bayesian methods were chosen to confirm HIV-2 groups and to investigate the likely origin of the sequences. Sixteen pol gene sequences were classified as group A, whereas one sequence was classified as group B. These phylogenetic relationships were supported by the posterior probability, which produced values ranging from 98% to 100% for the different groups (Fig. 1), and by the zero branch length test (p<0.001). The Bayesian tree showed that our strains are aggregated in several clusters depending by the country of origin and/or infection. Three patients (labeled as 1AA, 2PA, and 3SJ) clustered together, with 2PA and 3SJ closely related. All these patients seem to have an African origin and 2PA and 3SJ are somewhat related to sequences from Ghana, Ivory Coast, and Gambia. Interestingly, this clade also contains a sequence from a patient in Germany (labeled as BEN_M300502) who was believed to have been infected in Mali. Sequences from one patient (labeled as 4BR 1 and 2) appeared distantly related to a reference sequence isolated from Guinea-Bissau, whereas all four Indian sequences grouped together with other Indian isolates (Fig. 1). The sequence labeled as 10FO coming from Cape Verde grouped with sequences from Guinea-Bissau and/or Senegal.
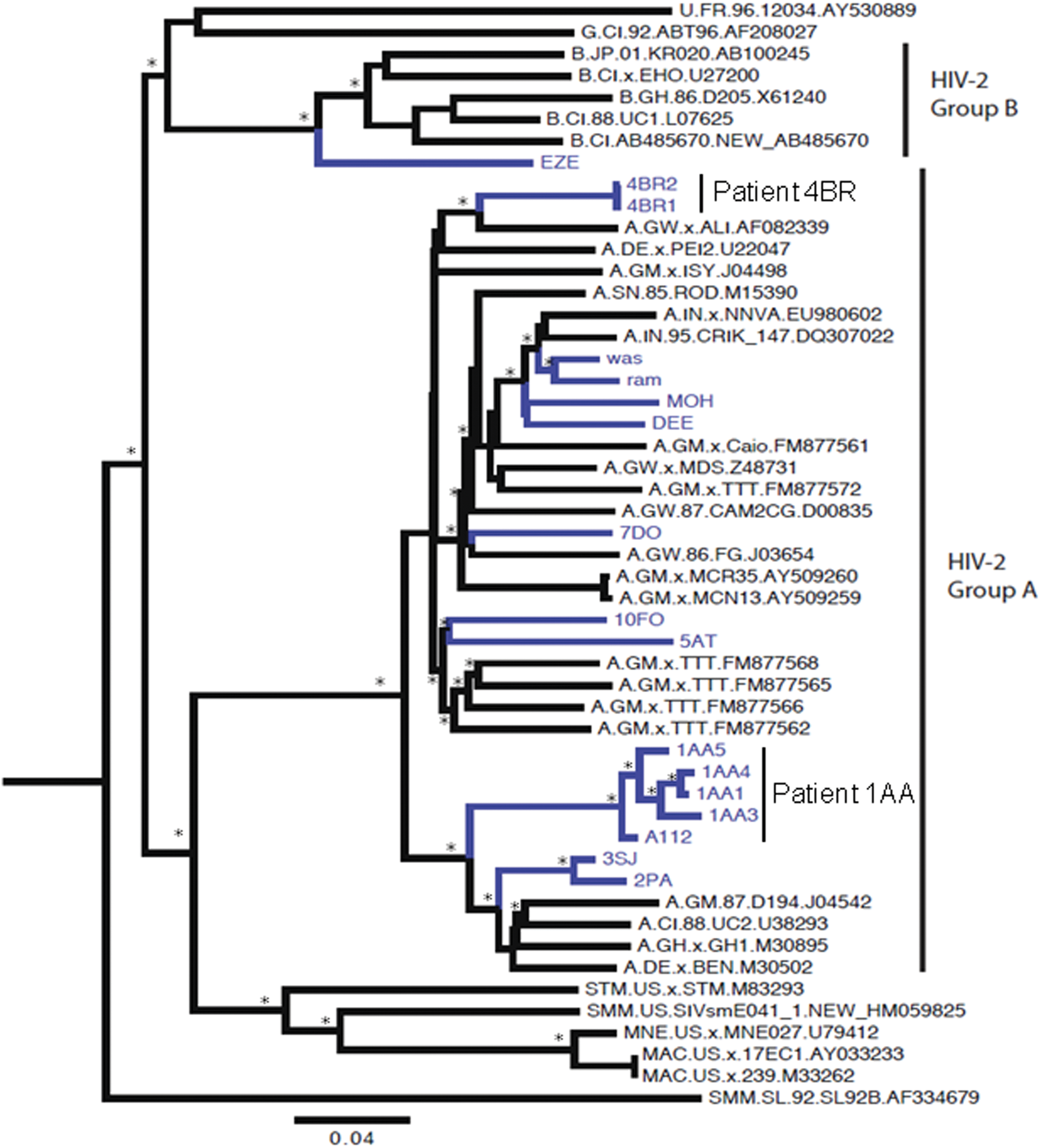
Bayesian phylogenetic tree of 17 HIV-2 pol gene (PR-RT) sequences plus 34 reference sequences downloaded from the HIV Los Alamos database (
Interestingly, phylogenetic analysis classified the pol gene sequences of one patient (labeled as 1AA) as a “non pure” group A because was in a marginal position within the cluster (Fig. 1). The env gene analysis revealed the virus from the same patient as group B (Fig. 2). Unfortunately SimPlot and split decomposition analyses did not reveal this particular recombination because the break point is located in an unsequenced region of the genome.
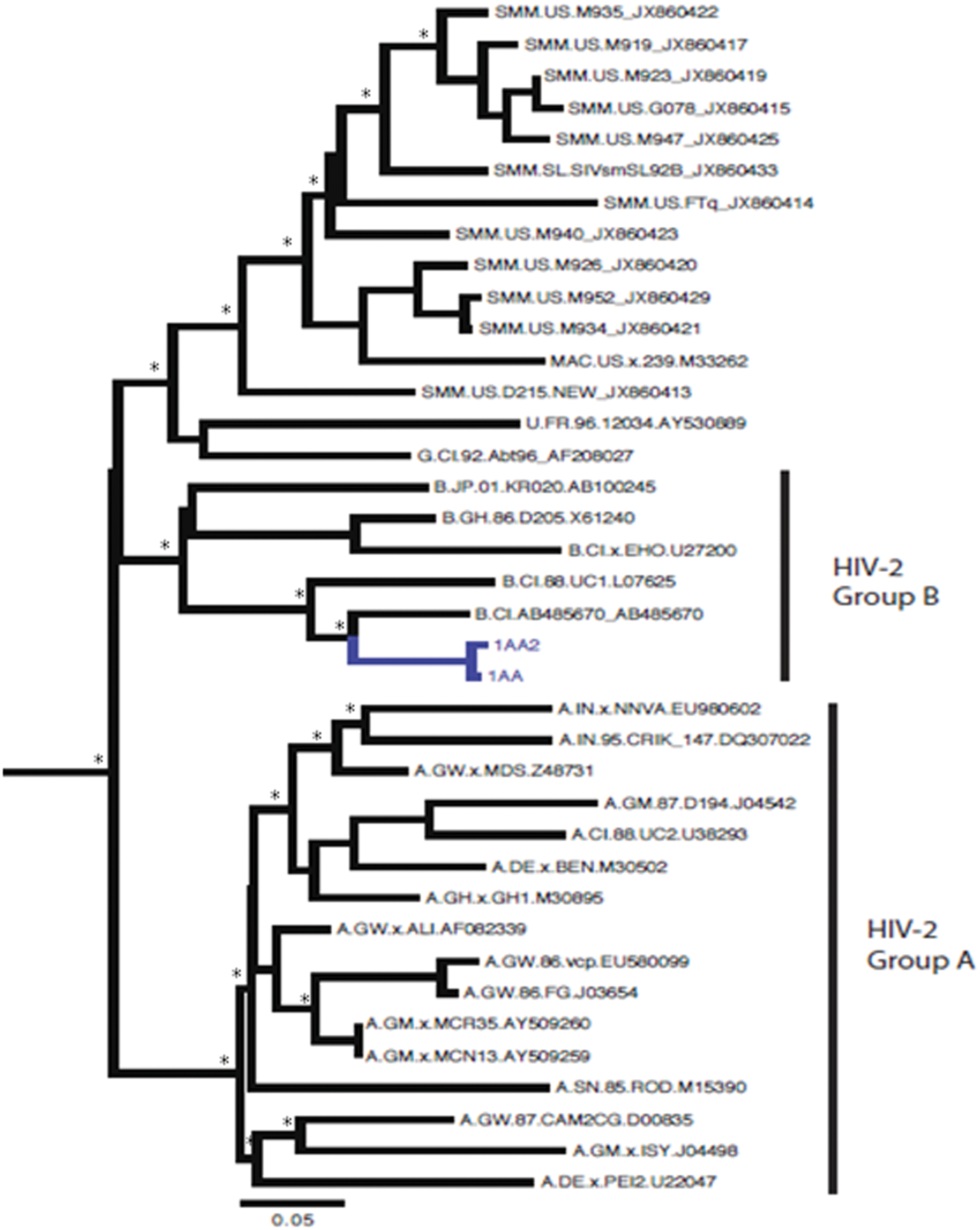
Bayesian phylogenetic tree of two HIV-2 envelope glycoprotein (env) gene sequences plus 36 reference sequences downloaded from the HIV Los Alamos database (
Discussion
HIV-2 infection is restricted primarily to West Africa, although the prevalence of HIV-2 is a growing concern in certain parts of Europe 14 and in the southwestern region of India. The highest prevalence (up to 8±10%) is encountered in Guinea-Bissau, a former Portuguese colony, 15,16 whereas the lowest prevalence of 1±2% is found in surrounding countries such as Gambia, Senegal, and Guinea. 17,18 Because of commercial sex workers, Gambia has the highest prevalence among these latter countries. In Europe, Portugal has the highest prevalence of HIV-2 infection 7,19 as well as countries with past socioeconomical links to Portugal including southwest India. 20 –22 Subtype A accounts for the majority of HIV-2 infections and is the predominant genotype in Guinea-Bissau and Europe. 23,24
In Italy, attention regarding this neglected infection increased recently because of the high prevalence of HIV-2 infection among immigrants coming from endemic areas. 25 To our knowledge, there are no updated estimates of HIV-2 infection in Italy, and only one paper by Costarelli et al. reports their own experience about the screening and management of HIV-2-infected individuals in Northern Italy. 25
Here we described 12 HIV-2 cases coming from different geographic regions but all registered in Italian medical care centers. A phylogenetic analysis was carried out to classify these 12 HIV-2 strains. The unique sequence classified as group B seems to be closely related to sequences from Ivory Coast and/or France. The Indian clade showed sequences that seem to be correlated with other HIV-2 infections in India. Considering the geographic position, the Cape Verde sequence is somewhat related to the Senegal and Guinea-Bissau sequences as expected. The two Burkina Faso sequences are closely related because the wife and husband had heterosexual transmission as a risk factor and a potential source of their infection seems to be Ghana, even though they cluster with sequences from other African countries such as Ivory Coast and Gambia. Interestingly, in this clade, as progenitor of the group we found a sequence (labeled as BEN.M30502) from a German AIDS patient who was probably infected in Mali. 26
Also in this interesting clade, there is a sequence belonging to a patient from Ivory Coast (labeled as 1AA) and classified as a probable “unique recombinant form.” This patient has bisexual transmission as the risk factor and it is possible that he is dually infected with HIV-2A and HIV-2B, but it is more likely that he is infected with an HIV-2A pol/B env recombinant because we did not detect HIV-2 B in the pol gene or HIV_2A in the env gene.
Two patients (labeled as 4BR and 5AT) with different sources of infection, both females and with different risk factors (transfusion and heterosexual transmission, respectively), showed no significant similarity to any database sequences.
The sequence from the patient from Guinea-Bissau labeled as 7DO showed a different topology in ML and Bayesian trees but in any case Africa seems to be his source of infection. It is well known that phylogeny cannot indicate the direction of virus travel, or who infected who without epidemiological information.
A surveillance system for HIV-2 is becoming critical because of the increase in migration from endemic areas to Italy. A correct diagnosis of HIV-2 infection is essential for planning the best therapeutic treatment. Therefore it is important to use valid diagnostic tools, i.e., antibody and Western blotting assays able to distinguish between HIV-1 and HIV-2 infection and sensitive and specific real-time PCR assays to quantities of the HIV-2 viral load. Phylogenetic analysis of the HIV-2 strains identified in the infected patients can help to trace the origin of the infection and the spread of the virus in the world.
Footnotes
Author Disclosure Statement
No competing financial interests exist.
Appendix
Members of the HIV-2 Study Group: G. Ceccarelli, Department of Public Health and Infectious Diseases, Sapienza University of Rome; E.N. Cavallari Department of Public Health and Infectious Diseases, Sapienza University of Rome; J.K. Maniar, Jaslok Hospital and Research Centre, Research Centre, Mumbai, India; V. Borghi, Clinic of Infectious and Tropical Diseases, University of Modena and Reggio Emilia; I. Bon, Department of Experimental, Diagnostic and Specialty Medicine, School of Medicine, University of Bologna; E. Chiari, Department of Infectious Diseases “Spedali Civili Di Brescia”; G. D'Offizi, I.N.M.I. “L. Spallanzani”.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.