
Abstract
Transmitted drug resistance (TDR) influencing nonnucleoside reverse transcriptase inhibitor (NNRTI) activity is increasing among new HIV-1 patients in several countries. As we recently observed an increase of K103N prevalence among new diagnoses in Belgium, we mined the Belgian national sequence database for homologous sequences. The earliest reverse transcriptase (RT) sequences available for drug-naive patients as well as sequences related to treatment failure were included. Fifty-five sequences were aligned and subjected to phylogenetic analysis, revealing the presence of a cluster of 29 virus sequences. All except one of those sequences were from antiretroviral (ARV)-naive patients at the time of sampling, and 22 had the K103N mutation. Epidemiological data of clustered patients were collected through the Institute of Public Health. Seventy-two percent of the clustered patients were infected through homosexual or bisexual contacts while the others reported heterosexual contacts only. All patients reside and were infected in Belgium. Sixteen were diagnosed between January 2011 and June 2012; 14 were aged between 18 and 29 years at the time of diagnosis. Nearly 60% of the clustered patients live close to the city of Namur, where HIV incidence substantially increased in the past 2 years. The identification of this transmission network advocates for local prevention reinforcement and underscores the need for continuous TDR monitoring. The spread of NNRTI TDR could affect ARV initiation schemes and prophylaxis strategies.
T
HIV-1 sequences were generated through routine clinical activity. Population sequencing was performed on viral RNA extracted from 1 ml of plasma using the Nuclisens Magnetic Extraction kit (Biomérieux, Marcy-l'Etoile, France). Protease (PR) and reverse transcriptase (RT) amplification and sequencing were done using the TRUGENE HIV-1 Genotyping Kit (Siemens Healthcare Diagnostics, Tarrytown, NY) on the OpenGene system. Sequences were assembled in the Integrated Database Network System (IDNS) from Smartgene (Zug, Switzerland).
During the second half of 2011, we observed several subtype B strains sharing more than 98% identity in the RT and showing the RT K103N mutation as well as the polymorphic PR C67S and V77I mutations. We mined both our local and the national IDNS database (ARLNET+) for related sequences with more than 95% identity as well as subtype B sequences with the K103N mutation in the RT. No distinction was made during that selection between ARV-naive and ARV-experienced patients. When several sequences were available for the same patient, only the oldest one was retained for the analysis. The three most recent sequences available from subtype C with the K103N mutation were selected for use as an outgroup in the phylogenetic analysis. Fifty-five PR and RT sequences were aligned separately with the MUSCLE software. 12 The RT codon 103 was omitted from the sequences to avoid a possible selection bias during phylogenetic analysis.
Phylogenetic trees were created by maximum likelihood 13 as implemented by the program PhyML version 2.4.5, 14 and by Bayesian analysis using MrBAYES version 3.1.2. 15 The general time reversible (GTR) model of evolution 16 was selected as the best model. The optimal parameters for the GTR model were estimated using the PhyML program. This model extended with gamma-shaped rate variation with four rate categories was used for both maximum likelihood (1,000 and 100 bootstrap samplings in PhyML for RT and PR, respectively) and Bayesian analysis (MrBAYES). To estimate Bayesian posterior probabilities, Markov chain Monte Carlo (MCMC) models were run for 1,500,000 (RT) or 1,000,000 (PR) generations and sampled every 100 generations [burn-in: 4,000 (RT) and 2,500 (PR) generations].
The phylogenetic analysis revealed the presence of one large cluster of 29 sequences, subdivided into two subclusters of 22 and seven sequences (Fig. 1). Probability support was 1, 0.89, and 0.98, respectively, and additional analysis performed either on the PR with the same model or with the maximum likelihood methodology for both targets revealed the same tree topology and clustering (data not shown). The only sequence among those 29 related to an ARV-experienced patient was sampled in 2003 (RT_146 in Fig. 1); virological failure with an NNRTI-including regimen was documented at that time. The 22 sequences in the largest cluster all harbored the K103N mutation, while the K103N mutation was absent in the seven samples of the smallest cluster.
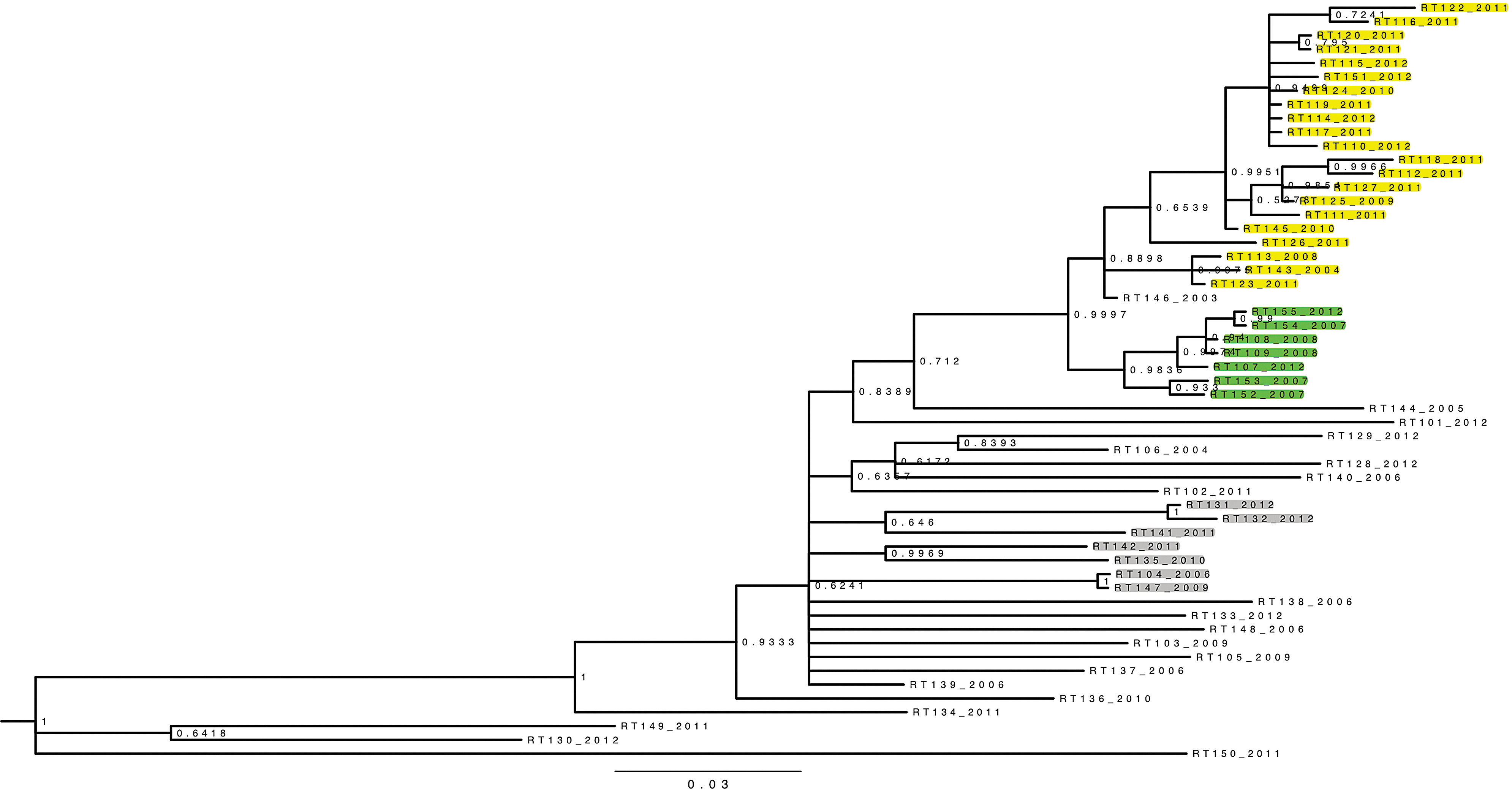
Phylogenetic analysis. Fifty-five reverse transcriptase (RT) sequences were aligned and analyzed omitting the codon 103 using Bayesian inference with the general time reversible (GTR) evolutionary model. Bayesian posterior probabilities are given at the nodes. The year identifying each sequence indicates the time of sampling, referring to the oldest sample available for each patient. All sequences aligned here have the RT K103N mutation except the seven sequences highlighted in green. All highlighted sequences (in yellow, green, and gray) are related to newly diagnosed antiretroviral (ARV)-naive individuals. RT146_2003 is the sole sequence of the large cluster related to an ARV-experienced patient.
Subsequently the information obtained through phylogenetic analysis was linked with the patients' epidemiological data (Table 1). The transmission network involved mainly men from Belgian nationality, and no case was related to contamination abroad. Sixty-five percent of patients were infected through homosexual contact while 28% reported heterosexual contacts only and 7% had partners from both genders. The median age at the time of infection was 24.5 years (18–52 years). Thirteen out of 22 sequences in the largest subcluster corresponded to patients living in or close to the city of Namur, while all seven sequences in the smallest subcluster were linked to patients living in the provinces of East and West Vlaanderen. Moreover, among those 22 sequences, a subcluster of 11 sequences can be distinguished (probability 0.95): all are related to recent infections, and also include all the cases of heterosexual transmissions. Figure 2 shows the number of new cluster members over time per province of residence, illustrating that the incidence of the K103N strain rose dramatically in 2011. Data for the year 2012 are not yet fully available, but the incidence for the first half-year remained high. Sixteen (55.17%) of the patients with the clustered viruses were diagnosed between January 2011 and July 2012, and 14 out of 16 were young adults aged between 18 and 29 years at the time of diagnosis.

Number of incident infections per year and per province of residence (Belgium) for the 29 patients involved in the transmission network. *First half of 2012.
National data were mined to establish the number of new diagnosis per year and per province: 15% of all HIV diagnosis in the province of Namur in 2011 were linked to the sole K103N strain described here. As a comparison, the prevalence of mutations linked to resistance (all ARV drug classes) in newly diagnosed patients in Belgium was 11% in 2009, which is close to the current European mean. 1 The analysis of new HIV cases in Belgium per province showed a clear rise in Namur since 2011, the province where the incidence was usually lower than the national mean. We suspect an outbreak rather than an increase of testing because the systematic genotyping of new cases was already introduced in 2009, five out of the 11 cases in the year 2011 had evidence of seroconversion, and the most probable place and year of infection were discussed with the patients related to the cluster. Furthermore, the incidence was probably underestimated as some cases are missing from our analysis: positive partners of subjects included in this transmission network died or were lost to follow-up, with no virus genotype available.
Those results plead for a reinforced prevention in that area. The start of ARV therapy in patients suspected of risky behavior, regardless of biological parameters, is also an option.
The observation of TDR involving the K103N mutation is not new, but the size of this cluster and its high incidence at a local level raise several issues. First, it supports the hypothesis that this mutation has no impact on transmission fitness. Although it is most likely that the K103N strain was initially selected in a patient during virological failure, the results of the phylogenetic analysis provide clear evidence for an ongoing transmission through different chains of infection in the absence of any drug pressure. Second, this finding shows that national or European global trends do not always fit with local epidemics, emphasizing the importance of continuous TDR and epidemiological surveillance. Even if men who have sex with men (MSM) and sub-Saharan African migrants are the most represented risk groups in Belgium, heterosexual transmissions in young adults were frequent in the cluster studied here. Third, in countries where ARV rollout was recent, NNRTI resistance is rising, advocating for the implementation of TDR surveillance systems.
The presence of K103N may also indicate the original presence of other NNRTI mutations that reverted over time. Care should be taken to use second-generation NNRTIs when the K103N mutation is detected in a newly diagnosed individual. Deep sequencing might be indicated in the future for those cases.
A global phylogenetic analysis of new HIV diagnoses at the national or supranational level would help to better understand the dynamic and fitness of transmitted strains and to describe the genetic particularities of the locally transmitted viruses. 17,18 In addition to the current epidemiological surveillance of HIV epidemics, phylogenetic data make it possible to link cases involved in chains of transmission and to investigate pathways and networks contributing to progression of an epidemic or an outbreak. Nevertheless, phylogeny should be used with caution: divergent genomes can exclude a direct link between two strains, but close proximity does not prove a direct transmission between individuals. The risk of data misuse should be kept in mind and data must always be treated anonymously.
Finally, knowledge of the most prevalent transmitted drug resistances can have an impact on ARV therapy initiation schemes as well as on the choice of drug combination used as preexposure or postexposure prophylaxis, at least in countries where it is available.
Footnotes
Acknowledgments
The authors warmly acknowledge Professor F. Opperdoes, retired from the de Duve Institute, Université Catholique de Louvain (Brussels, Belgium), for his advice on and support of the phylogenetic analyses. We also thank all the Belgian AIDS reference laboratories staff and clinicians involved in data collection, not cited here for patient confidentiality reasons.
The project was approved by the CEBHF (Comission d'Ethique Biomédicale Hospitalo-Facultaire) of the Université Catholique de Louvain, Brussels, Belgium.
J.R. supervised the project, analyzed the data, and drafted the manuscript. M.G.I. collected virological and medical data. K.J. performed sequence alignments and phylogenetic analysis. N.A. and A.V. validated the data with regard to medical records. A.S. provided the epidemiological data at the national level. P.G. directed the project and all the authors revised the manuscript.
The present work was partially presented as a poster at the 11th International Congress on Drug Therapy in HIV infection, 11–15 November 2012, Glasgow, UK.
Author Disclosure Statement
No competing financial interests exist.