
Abstract
HIV-1 epidemics are expanding among men who have sex with men in low- and middle-income countries. To confirm and further explore preliminary data in Senegal, we aimed to determine 3 years after a first study the HIV-1 genetic diversity in three different viral regions. From 109 samples available in 2007, 93 were sequenced in gag, 77 in env, and 60 in pol. Phylogenetic analysis showed that subtype C predominated (38–52%), followed by CRF02_AG (30–40%), subtype B (13–17%), and CRF09_cpx (2.6–5%). Subsubtype A3 and strains tightly linked to CRF43_02G were identified in env and gag, respectively, and 12% of the samples were unique recombinants. Six transmission chains involving two to seven individuals were identified. Some strains carried resistance mutations inside transmission chains. This study confirmed the existence of a dual epidemic in Senegal and emphasized the need to strengthen prevention programs to avoid strains intermixing between low-risk women and high-risk men.
A
The first report on the genetic diversity in the MSM population in Senegal was realized in patients sampled in 2004. Based on partial pol sequences only, the predominance of subtype C (40%) followed by CRF02_AG (28%) and subtype B (19%) was shown. 5 These data were different from those found in the general population 6,7 as well as in female sex workers (FSW) 8 in Senegal, where CRF02_AG strains were predominant (64%), but many other circulating strains were found (A1, A3, B, C, D, G, CRF06_cpx, CRF09_cpx, and CRF45_cpx). In female sex workers, the HIV-1 subtype/CRF distribution was comparable to that found in the general population; however, subsubtype A3 has become more prevalent over time in this population group. 8 The prevalence of subtype C strains in Senegalese MSM was intriguing because these levels of this subtype have never been observed in West Africa. 9 A recent study investigated the evolutionary history of this subtype, which predominates today in the MSM population. 10 These analyses showed a significantly well-supported cluster, which contained all subtype C strains that circulate among MSM in Senegal, and demonstrated that in contrast to southern African countries, subtype C did not become the predominant strain in Senegal and spread efficiently only in the MSM population group, as the result of a single introduction.
Our aim was to investigate in more detail the genetic diversity in the MSM population from Senegal 3 years after the first study, to evaluate whether the HIV-1 subtype/CRF distribution was stable over time. To this end, three different genomic regions (gag p24, pol protease+reverse transcriptase, and env V3-V5) were analyzed.
During the epidemiological survey in 2007, 109 HIV-1-infected MSM were identified out of 500 anonymously recruited men in four urban sites throughout Senegal. The general characteristics of the whole MSM cohort and factors associated with HIV infection have been previously described. 11 Our study focused on 97 individuals from whom at least one HIV-1 genomic region was amplified. The sociodemographic characteristics of the study population are given in Table 1. The patients were recruited from four urban sites throughout Senegal: the capital city Dakar (n=81) and three other major cities at 70 to 260 km distance, i.e., Mbour (n=6), Saint-Louis (n=4), and Thies (n=6). The studied patients were mainly Senegalese (97.9%), unmarried (90.6%), and young; 68% of them were less than 30 years old (median age 26 years old ranging from 18 to 56 years old). Sixty-four (68.82%) MSM had regular male partners; however, most MSM reported having had additional partners during the past 12 months: 90 (97.8%), 23 (25%), 24 (26.08%), 10 (10.87%), 11 (11.96%), and 4 (4.35%) reported Senegalese partners, with others reporting African partners and European, American, Lebanese, or Asian male partners, respectively. Interestingly, 41.5% of MSM (39/94) declared that they had at least one regular female partner, 66.3% (61/92) reported at least one heterosexual contact during the past 12 months, and one-third declared that they participated in the first 2004 MSM survey. In addition, nine patients were on first-line antiretroviral treatment.
Blood samples were collected in EDTA tubes, and buffy coats were isolated and stored at −20°C. DNA was then extracted with a QIAamp Viral DNA kit (Qiagen, Courtaboeuf, France) and genetic characterization in gag and env genes was performed by direct sequencing of the p24 region and the V3-V5 region, respectively, as previously described.
6
For the pol gene, genetic characterization was performed on DNA extraction by direct sequencing of the full protease (PR) and partial reverse transcriptase (RT) with two nested polymerase chain reactions (PCR), generating overlapping segments, using the primers in the ANRS AC11 procedure without reverse transcription. (
Out of the 109 samples, 93 could be amplified and sequenced in the gag p24 region, 77 in the env V3-V5 region, and 60 in pol (PR+RT); 27 were sequenced in two genomic regions and 53 in the three regions. The newly generated sequences were submitted to the EMBL database under accession numbers HF947599 to HF947658, HF947659 to HF947751, and HF947752 to HF947828 for the full PR and partial RT, p24 protein region, and env V3-V5 region, respectively.
The newly determined sequences were aligned with known representatives of the different subtypes/circulating recombinant forms (CRFs) using MUSCLE as implemented in Seaview version 4 (
The subtype/CRF distribution of HIV-1 strains in the three genomic regions (Table 2) was obtained by combining the results from individual phylogenetic trees and from bootscan analysis. Subtype C strains were predominant with a prevalence of 38.3%, 43%, and 52% in pol, gag, and env, respectively, followed by CRF02_AG strains (30–40%) and then subtype B (13.3–17%). The CRF09_cpx virus represented 2.6–5% of the circulating strains, subsubtype A3 was detected in the env gene only, and CRF43_02G strains in the gag gene only. A3 was found in one strain that was CRF02_AG in gag and pol, and CRF43_02G was identified in gag in two strains that could not be amplified in the other regions. To our knowledge, this is the first report of CRF43_02G strains in Senegal. These viruses, composed of subtype G in gag and the first part of pol, were characterized in 2008 12 and could have been missed by the first analysis on the genetic diversity in MSM, due to the absence of reference strains at that time. As some subtype G strains had been found in the MSM population group sampled in 2004, 5 these sequences were reanalyzed to determine if they belonged to the CRF43_02G clade. SimPlot analyses showed that those strains were globally subtype G, but most of the pol fragment was very close to the subtype G constituting the CRF43_02G strains (Fig. 1). Such subtype G strains might be one of the parents of the CRF43_02G strains.

Bootscan and phylogenetic tree analysis of men who have sex with men (MSM) subtype G pol sequences sampled in 2004. Top: Bootscan plot performed under SimPlot software as indicated in Materials and Methods. Bottom: Maximum likelihood phylogenetic tree for each segment as previously defined in the bootscan analysis.
CRF, circulating recombinant forms; URF, unique recombinant forms; nd, not determined.
Bootscanning analyses found a 3.3% and 2.1% prevalence of unique recombinants in the pol and gag regions, respectively. In the gag p24 region two strains had a significant central breakpoint: the first one between subsubtype A3 and CRF02_AG and the second one between subtypes C and D (Fig. 2a). In the pol region, two unique recombinants, CRF02_AG/C and C/CRF02_AG, were identified (Fig. 2b). However, pol sequences were first generated with the ANRS method, producing two overlapping fragments for the protease and for the reverse transcriptase, which were assembled as a consensus sequence for the whole segment (PR+RT). Recombination analysis on this first set of sequences showed a total of five unique recombinants out of a total of 60 sequences.
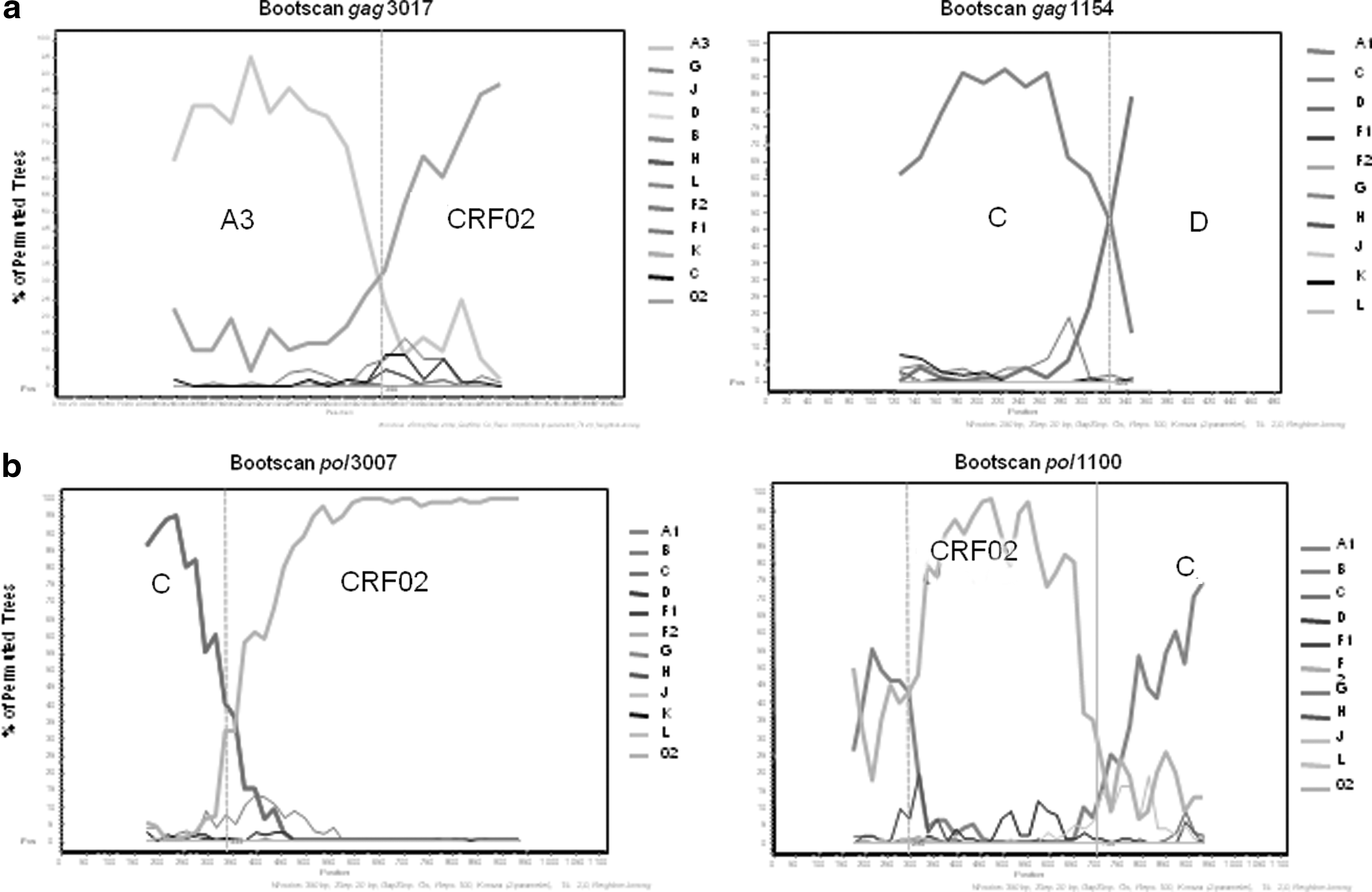
Recombination profiles of MSM strains. Recombination profiles of two strains in the gag p24 region
The same samples were reamplified directly as a single fragment (1,140 nucleotides) by using an in-house method. In that case, three sequences were revealed to be nonrecombinant: two subtype C samples initially classified as URF (CRF02_AG/C) from which the protease fragment, identified as CRF02_AG, was phylogenetically different between them and also from the other CRF02_AG sequences of the present study, and a third subtype C sample previously classified as unique recombinant C/F/D. The subtype discordance obtained when using different PCR methods raises the possibility of dual infections with more than one HIV-1 strain. High rates of dual infections (up to 20%) were already reported by using the multiregion hybridization assay (MHA) in high-risk patients from East Africa, India, and South America. 13 –15 To date, no data have been reported in West and Central Africa, because of the high genetic diversity of HIV-1 in this geographic area. 9 The present study clearly indicated that at least some MSM may be infected by more than one virus, and that viral loads may be high as different viruses were obtained by using different PCR conditions.
Sequencing three different regions made it possible to investigate the prevalence of interregion recombinants. Globally, 53 samples were analyzed in the three targeted regions, 27 in two regions, and 17 in one region only (14 samples in gag, one sample in pol, and two samples in env). On 80 samples that were sequenced in more than one region, seven samples for which the subtype designation was different were identified: one gagC/envCRF09_cpx, one gagCRF02_AG/polC, one gag-polCRF02_AG/envA3, two gag-polCRF02_AG/envC, and two gag-polCRF09_cpx/envCRF02_AG. The prevalence of the interregion recombinants was therefore 8.75% (7/80). As the unique recombinants identified either in gag or pol and the above interregion recombinants were not from the same samples, we could evaluate the global prevalence of recombinant strains in the MSM population group as 11.34% (11/97). The prevalence of unique recombinant strains in the general population from Senegal was estimated at about 12%, 7 comparable to that found among MSM today. In the first genetic survey on Senegalese MSM a lower prevalence of unique recombinant forms (URFs) (4.3%) was seen, 5 due to the fact that only one viral region was studied.
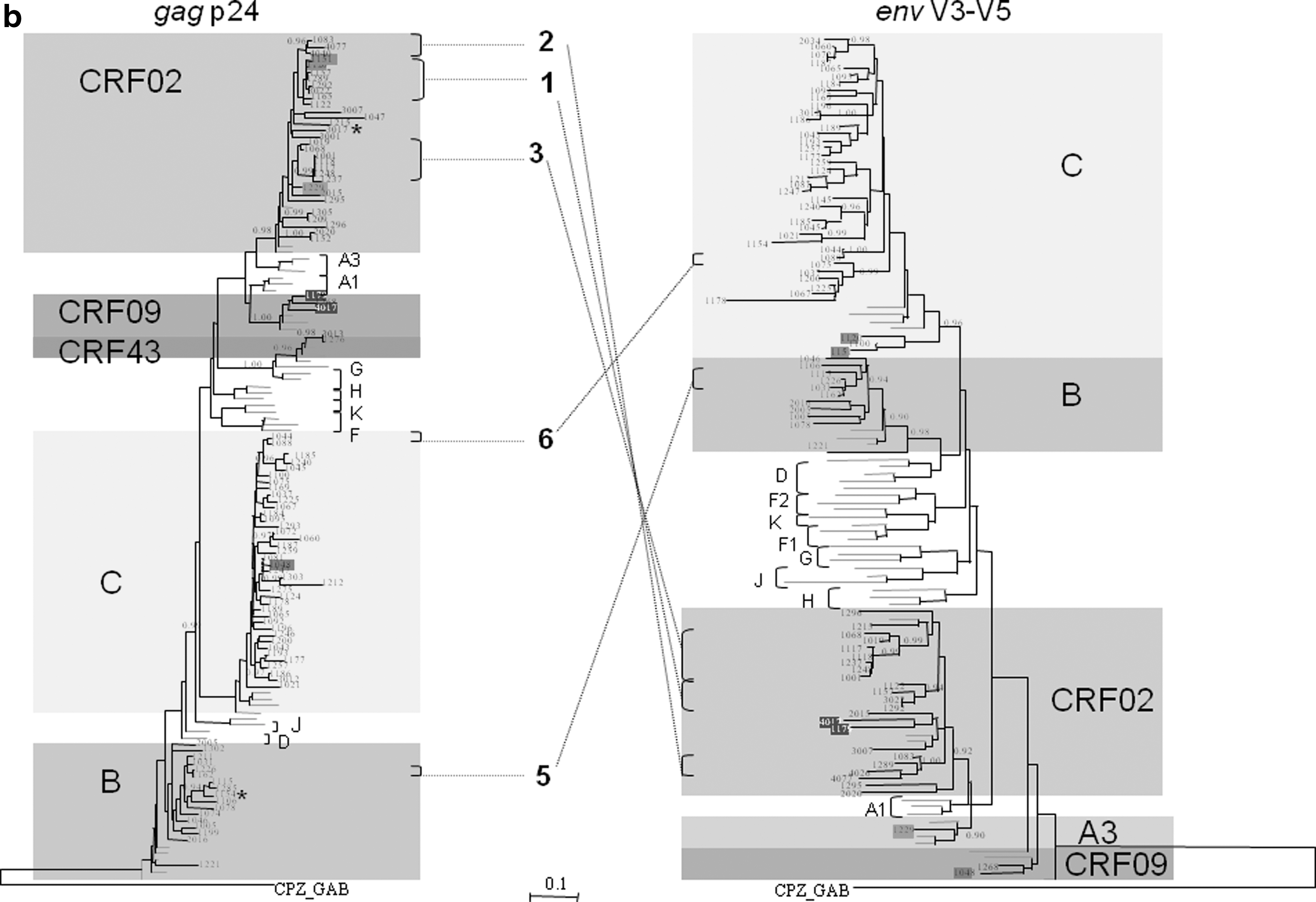
Figure 3 illustrates the phylogenetic relationships between the sequences of the present study in the three targeted regions. For better clarity, the general trees were drawn using the same approach as described above, with a minimal number of reference sequences, i.e., excluding those not represented among the samples of the present study.
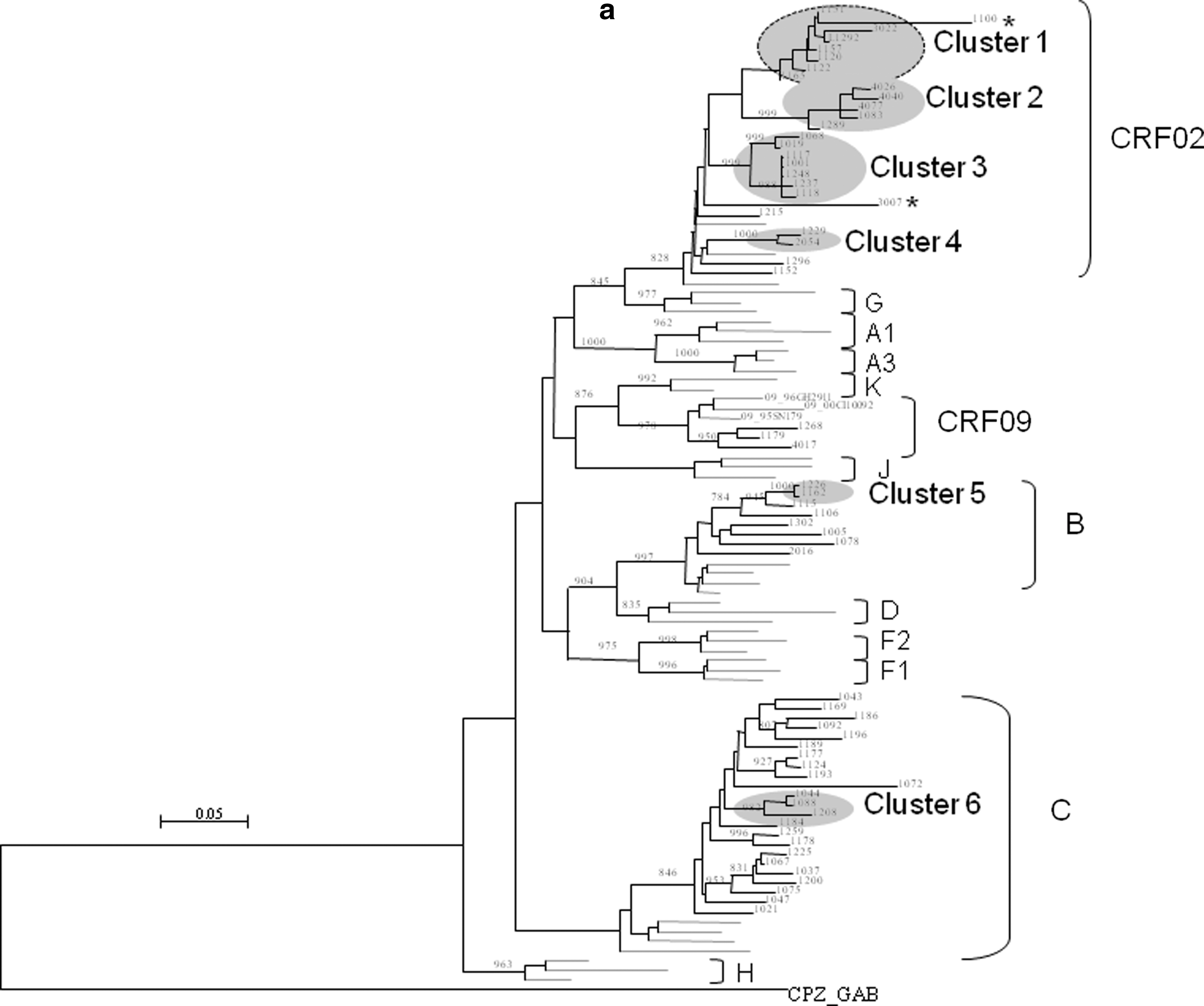
Maximum likelihood (ML) phylogenies of MSM strains in Senegal. Maximum likelihood phylogenetic trees for the pol PR-RT region
To better assess the phylogenetic relationships among 60 pol sequences, a maximum likelihood analysis with PhyML was performed using 1,000 bootstrap resamplings. The existence of transmission clusters was ascertained using the statistical robustness of the ML topologies based on high bootstrap values (98%) with 1,000 bootstrap resamplings and short branch lengths (≤0.015) in pol gene sequences, following the established criteria defined in previous studies. 5 Of 60 pol sequence samples analyzed, 26 (43.33%) sequences segregated into six transmission clusters as defined by PhyML (Fig. 3a). Four transmission clusters were found in the CRF02_AG clade; one was identified for subtype C and one for subtype B. The number of individuals varied from two to seven in the different clusters. Interestingly, MSM were from different localities in three transmission chains: in cluster 1 with eight sequences, one MSM was from Mbour and the others from Dakar (85 km distance); in cluster 2 (n=5 sequences), two MSM were from Dakar and three from Saint-Louis (260 km distance); and in cluster 4 (n=2 sequences), one MSM was from Dakar and one was from Thies (70 km distance). Contact tracing was not possible to confirm the transmission chains, because anonymity was required in the epidemiological survey in order to protect individual identities.
A unique recombinant CRF02_AG/C segregated inside cluster 1 (Fig. 3a) because the phylogenetic signal was mainly from CRF02_AG. When the tree was drawn without the URF sequence, the bootstrap value for cluster 1 was 1,000 and the mean branch length fall to 0.00878, both following the criteria necessary to define a transmission chain. Therefore the cluster was illustrated as in Fig. 3a. The sequences that segregated into transmission clusters in pol were investigated to see whether they also clustered together in the gag and/or env genes. Results were presented in Table 3. Most strains that segregated into clusters in the pol region also clustered together in both the gag (21/26=80.8%) and env (19/26=73.1%) genes. Only one sample was positioned inside the same clade but not in the cluster in gag. Figure 3b illustrates the cluster distribution in the gag and env genes, relative to the transmission chains identified in pol. Globally, PhyML analysis in the pol gene showed that 26/60 (43.3%) strains segregated into transmission clusters in 2007, versus 67% (47/70) in the 2004 survey. 5
Same samples in gag and env.
n, number of strains; REC, interregion recombinant strain; ns, not sequenced.
The transmission chains were observed mainly among CRF02_AG strains (21/26, 80.7%), whereas they were regularly distributed among the different clades in 2004. Notably, subtype C sequences that segregated in 2004 represented nearly 45% (21/47) of the strains clustering into transmission chains, versus 11.5% (3/26) in 2007. As already stated, clustered transmissions are the driving force of the MSM epidemic, 16 and therefore they represent recent and homogeneous infections. Their decrease could therefore reflect a spread of the epidemic, which is no longer limited to a group of identifiable individuals. Alternatively, it may be possible that differences in the number of transmission chains are due to different samplings.
Generally, pol sequences from MSM were grouped separately from HIV-1 references in both subtype B, C, and CRF09_cpx clades (Fig. 3a). This did not seem to be the case for CRF02_AG strains, whose sequences from MSM and HIV-1 references were intermixed (Fig. 3a). The same was also observed in gag and env trees (Fig. 3b). The specific distribution of MSM subtype C sequences, sampled in 2004, relative to the subtype C strains from the general population has already been described. 10 MSM subtype C sequences from the actual survey were added to previous sequences from 2004 and with subtype C sequences from the general population of Senegal. This analysis confirmed the previously known distribution of subtype C strains from MSM compared to the sequences from the general population (not shown). The same analyses were attempted for CRF02_AG strains and subtype B and CRF09_cpx sequences. Although probably insufficient numbers of sequences were used, MSM CRF02_AG pol sequences from both surveys seemed intermixed with CRF02_AG strains from the Senegalese general population (not shown).
Finally, pol amino acid sequences were analyzed for the presence of mutations in the protease and reverse transcriptase genes. Four patients for whom pol sequences were available were under first-line antiretroviral treatment, therefore the amino acid sequences were investigated with the last updated online Stanford Resistance Database tool: the HIValg program, combining three resistance algorithms (ANRSV2011.05, HIVDBV6.2.0, and RegaV8.0.2). Two of them carried drug resistance mutations: one (1200) carried the mutations Y181C and H221HY and was resistant to the NNRTI drugs, although the sequence was separated from those of other MSM sequences in the phylogenetic tree; the second (2054) carried K101Q, K103N, V108I, and M184V and was resistant to the nonnucleoside reverse transcriptase inhibitors (NNRTIs) except rilvipirine and to lamivudine/emtricitabine (3TC/FTC). This second one was inside cluster number 4 with another MSM sequence from a different town (Fig. 2a). Because the remaining 56 pol sequences were from naive individuals, transmitted drug resistance (TDR) was evaluated through the CPR tool following the SDRM 2009 mutation list (ver.6.0
The aim of this study was to investigate, 3 years after the first survey, the genetic diversity of HIV-1 strains circulating in the MSM population group in Senegal. The objectives were to determine whether the specific pattern of strains circulating among MSM changed or did not change over time, to describe the genetic diversity in three viral regions instead of one, and to specify the prevalence of recombinant strains. Genetic subtyping confirmed the predominance of subtype C strains, comparable to that found in the earlier survey. 5 CRF02_AG and subtype B were again the second and third most represented HIV-1 clades circulating in the MSM population group, although they seemed to be less prevalent than 3 years ago.
As about one-third of the MSM declared that they had participated in the first survey in 2004 and because personal data were anonymous, it was not possible to directly compare the prevalence of the different strains between the two periods. However, as two-thirds of the MSM were newly recruited, identification of major changes in the subtype/CRF distribution between the two periods of time remained possible. Globally, the specific pattern of strains circulating among MSM did not change over time.
Analyzing three different genomic regions made it possible to assess the global prevalence of recombinant strains (11.8%), which were mainly composed of the subtypes and CRFs circulating among the MSM population group. However, some unique recombinants involved fragments of subtypes that were not yet reported among Senegalese MSM. More importantly, unique recombinants were identified or not identified depending on the pol PCR conditions, strongly suggesting that dual infections may occur in the Senegalese MSM population, and should be further investigated.
Globally, the study confirmed the existence of a dual epidemic in Senegal with a pattern of strains very different between the MSM and the general population groups. These analyses raised questions about the possible evolution of the transmission of HIV-1 strains within the MSM community, and also to or from the general population. The HIV-1 situation in Senegal can be described following a scenario in which MSM, intravenous drug use (IDU), and heterosexual transmission all contribute significantly to the HIV epidemic. 17 In this context, it is of interest to continue the surveillance of circulating strains and, more importantly, to further explore the relationships and the natural history of the viruses common to both populations. Finally, as some strains carried resistance mutations inside transmission chains, it remains crucial to develop and implement better targeted HIV prevention programs and interventions.
Footnotes
Acknowledgments
We give special thanks to the other professionals dedicated to patient care in the participating centers and the patients for their participation. This study was supported by MoH, through its division for HIV/AIDS and STI. We extend our gratitude to WANETAM for the additional support. The funders had no role in the study design, data collection and analysis, decision to publish, or preparation of the article.
Author Disclosure Statement
No competing financial interests exist.