
Abstract
An increasing number of circulating recombinant forms (CRFs) and unique recombinant forms (URFs) all over the world has necessitated being vigilant about new recombinants. Since the first report of a recombinant virus with an A1/C mosaic in 1998 more and more B/C and A/C recombinant viruses are being reported from India. Here we report the identification and characterization of a unique HIV-1 A1/C recombinant circulating in Western India. Analysis of the full-length genome using RIP, SimPlot, and jpHMM@Gobics has confirmed its mosaic structure with insertion of subtype A1 in the backbone of subtype C at three positions: gag-pol (1973±15–2617±47), pol-vif (4879±37–5582±32), and gp41 (8437±106–8811±8); however, RIP and SimPlot showed one more small insertion in integrase (4343–4519). Phylogenetic analysis confirmed that the recombinant virus has an insertion of clade A1 in the backbone of subtype C, which has come from Indian subtype C.
Introduction
M
A study of the intravenous drug user (IDU) cohort from Northeastern India showed the effectiveness of the Multiregion Hybridization Assay (MHA) for genotype-based surveillance, and along with full genome sequencing the assay can serve as an efficient tool for the characterization of a recombination pattern among the newly emerging HIV-1 recombinants. 4 Most of the other molecular characterization studies in India are based on sequencing of a part of the genome. Very few reports of a full genome sequence have emanated from India.
Six A1C recombinant viruses have been reported from India, out of which only two are characterized based on a full-length genome sequence 5,6 and four based on partial genome data from Calcutta, 7 Bengaluru, 8 and Pune. 9 B/C recombinant viruses were reported from Manipur by Lakhashe et al. 10,11 based on full-length genome sequencing and by Bhanja et al. 12 based on partial gene sequencing. B/C recombinant viruses were also reported from West Bangal, 13 Karnataka, 14 and Maharashtra based on partial genome sequence data. CRF02_AG has also been reported from Delhi. 15
Materials and Methods
As a part of the project “full-length genome analysis of HIV-1 isolates from India” a blood sample was collected from a 27-year-old male seropositive individual (NARI Patient Id: 09-387) having a CD4 count of 220/mm3 from Pune, India in July 2009. Peripheral blood mononuclear cells (PBMCs) were isolated by density gradient centrifugation using Ficoll-Hypaque (1.077 g/ml, H8889, Sigma, St. Louis, MO). Isolated PBMCs were cocultured for 21 days with an equal number of mitogen-activated (5 μg/ml of PHA-P, Sigma-Aldrich) PBMCs from a healthy HIV infection free individual. During the process both types of PBMCs were suspended in RPMI 1640 (Hi-Media) supplemented with 10% fetal bovine serum (FBS) (Sigma-Aldrich), 20 U/ml human interleukin-2 (Roche), 100 U/ml penicillin, 100 μg/ml streptomycin (Hi-Media), and 2 mM
The culture supernatant was tested for p24 antigen on days 7, 14, and 21 postinfection (PID) by HIV-1 antigen micro ELISA (Biomerieux). Proviral DNA was extracted from PBMCs from p24-positive culture using the QIAamp DNA blood mini kit (Qiagen, Hilden, Germany). Isolate NARI-FLS_09-384 was amplified with two fragment strategy with an overlap of 0.88 kb nucleotides. The 5′ terminal 2.88-kb fragment of the 5′ LTR through the RT region (0001–2878 in the HXB2 coordinate) was amplified by nested polymerase chain reaction (PCR) using outer primer pairs N558F and N453R in the first round and inner primer pairs N533F and N502R 16 in the second round, and a 7.5-kb fragment p7Gag through the 3′ LTR (1996–9632 in the HXB2 coordinate) was amplified using primer pairs Uninef7′ and pro5F. 17
Amplified products were cloned into a pCR2.1 vector and eight clones of 2.88 kb (0001–2878 in the HXB2 coordinate) and one clone of 7 kb (1996–9632 in the HXB2 coordinate) were generated. DNA sequencing of amplified products of both the fragments from proviral DNA and all the clones was done using an ABI PRISM Big Dye terminator v3.1 cycle sequencing kit (Applied Biosystems) with the help of an automated genetic analyzer ABI 3730XL DNA Sequencer (Applied Biosystems Inc., Foster City, CA). A total of 45 primers was used to cover the entire length of the viral genome and a contiguous sequence was made using the primer walking method with the help of Seqscape v2.6 (Applied Biosystems). A full-length genome sequence (NARI-FLS_09-387) of a 9615 nucleotide-long position (1–9603 relative to the HXB2 genome) and spanning 5′ LTR to 3′ LTR was obtained by sequencing the amplified product directly.
The full genome sequence NARI-FLS_09-387 obtained by sequencing the PCR product was aligned with HIV-1 strains of reference subtypes and subtype C from India obtained from the Los Alamos HIV Database (
Results
The intracluster average genetic distance of Indian clade C was 6.34%, the average genetic distance of NARI-FLS_09-387 with the Indian clade C group was 9.42%, while the subsubtype distances between B–D and F1–F2 were 10.7% and 11.2%, respectively. The sequence NARI-FLS_09-387 clustered with the clade C cluster but away from Indian subtype C and the distance from Indian clade C suggesting the possibility of a recombinant genome (Fig. 1).

Phylogenetic analysis of newly derived full genome sequences of recombinant isolates from India. The sequence of NARI-FLS_09-387 was aligned with Indian clade C (accession numbers AY049708, EF694036, EF469243, AF067154, AF286232, AB023804, AY713414, AF067157, AF067159) and reference sequences of all pure subtypes (A–K) available at the Los Alamos HIV database. The scale bar represents 2% genetic distance.
The full genome sequence was analyzed with RIP (

Identification of recombinant isolate 09-387. Similarity plots were drawn using RIP 3.0 software with window size: 300 nucleotides; confidence threshold: 90%; gap handling: strip all gaps and plot all window values; and multistate character: Yes. Similarity with subtypes C and A1 shows insertion of subtype A1 at four positions (denoted as 1, 2, 3 and 4) in the backbone of subtype C. Color images available online at
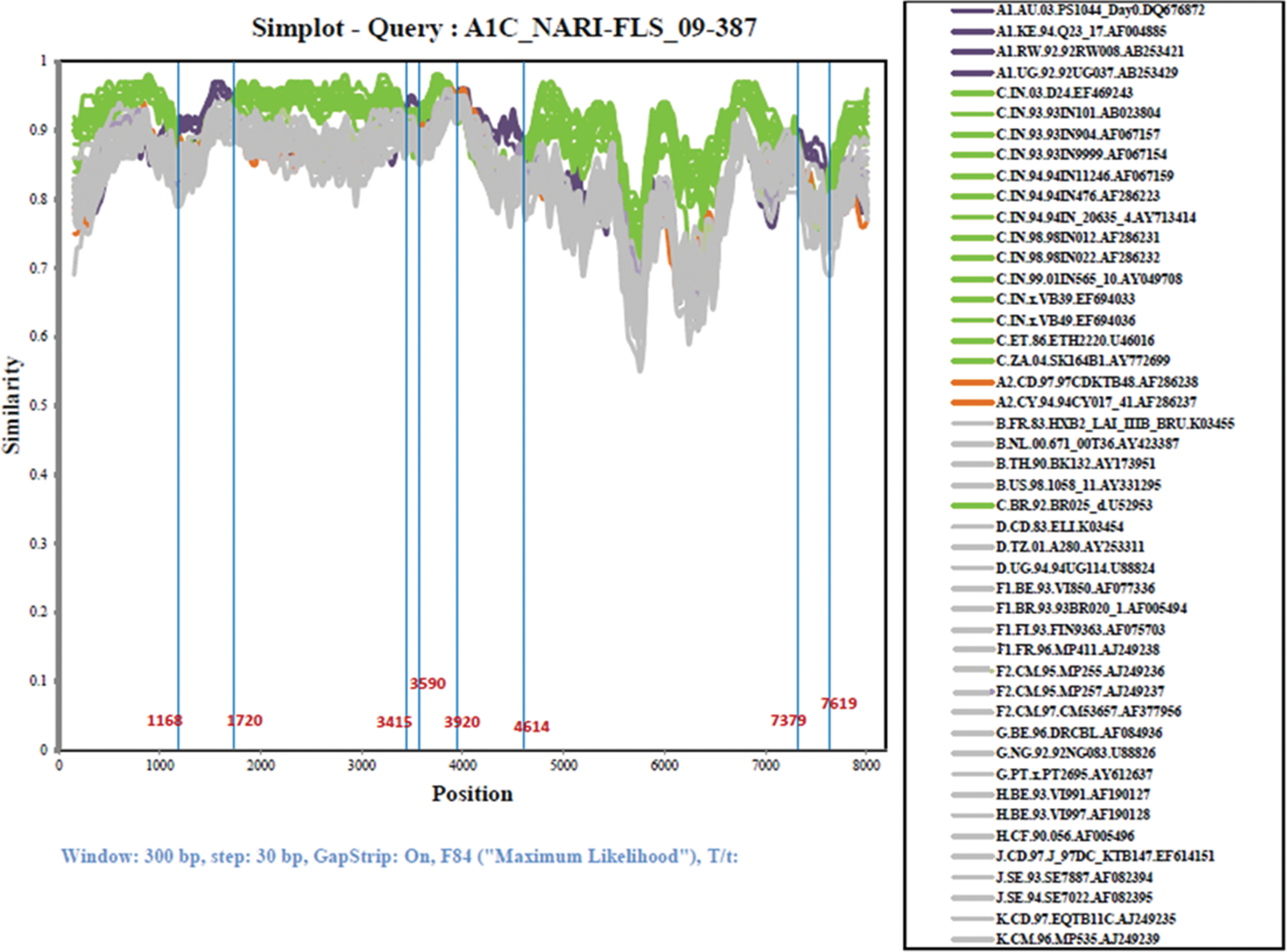
Subtype identities of isolate 09-387. The similarity plot was created using SimPlot 3.5.1 software configured with window size: 300 nucleotides; step: 30; GapStrip: On, F84 (maximum likelihood). Breakpoints are shown at the bottom of the plot, which indicates insertion at four places (nts 1168–1720, 3415–3590, 3920–4614, and 7379–7619). Reference sequences of the subsubtypes A1, A2, B, C, D, F1, F2, G, H, J, and K, as well as of subtype A1 and C from India, are listed on the right side. Color images available online at

Breakpoints in HXB2 coordinates.
Phylogenetic analysis was performed next using a full-length genome sequence obtained from PCR products based on the breakpoints received from jpHMM@Gobics and SimPlot to confirm potential ancestor subtypes from which the recombinant strain was derived. Segment II spanning p7gag-RT (1980–2600), IV spanning the end of IN-Vif (4850–5549), and VI spanning gp41 (8400–8800) genes clustered within clade A1 radiation with A1 isolates sequenced in the study with significant bootstrap value (≥90) (Fig. 6). The segment I Gag (791–1970), V Vif-Gp41 (5560–8430), and VII Gp41-Nef (8481–9400) clustered within clade C radiation along with Indian isolates but segment III Pol (2605–4845) clustered away from subtype C (Fig. 5) radiation suggesting the possibility of insertion of clade A1 and corroborating the insertion of clade A1 in the integrase gene (4343–4519 HXB2 coordinates) as revealed in SimPlot analysis.

Phylogenetic analysis based on breakpoints of segments showing subtype C. Each segment was analyzed separately by the phylogenetic maximum likelihood method under the General Time Reversible Model (GTR model) and the discrete Gamma was distributed with Invariant sites (G+I) with the number of discrete gamma category 5. The genetic distance corresponding to the lengths of the branches is shown by the bottom line. Segments I, V, and VII clustered within clade C radiation but segment III clustered away from subtype C radiation.
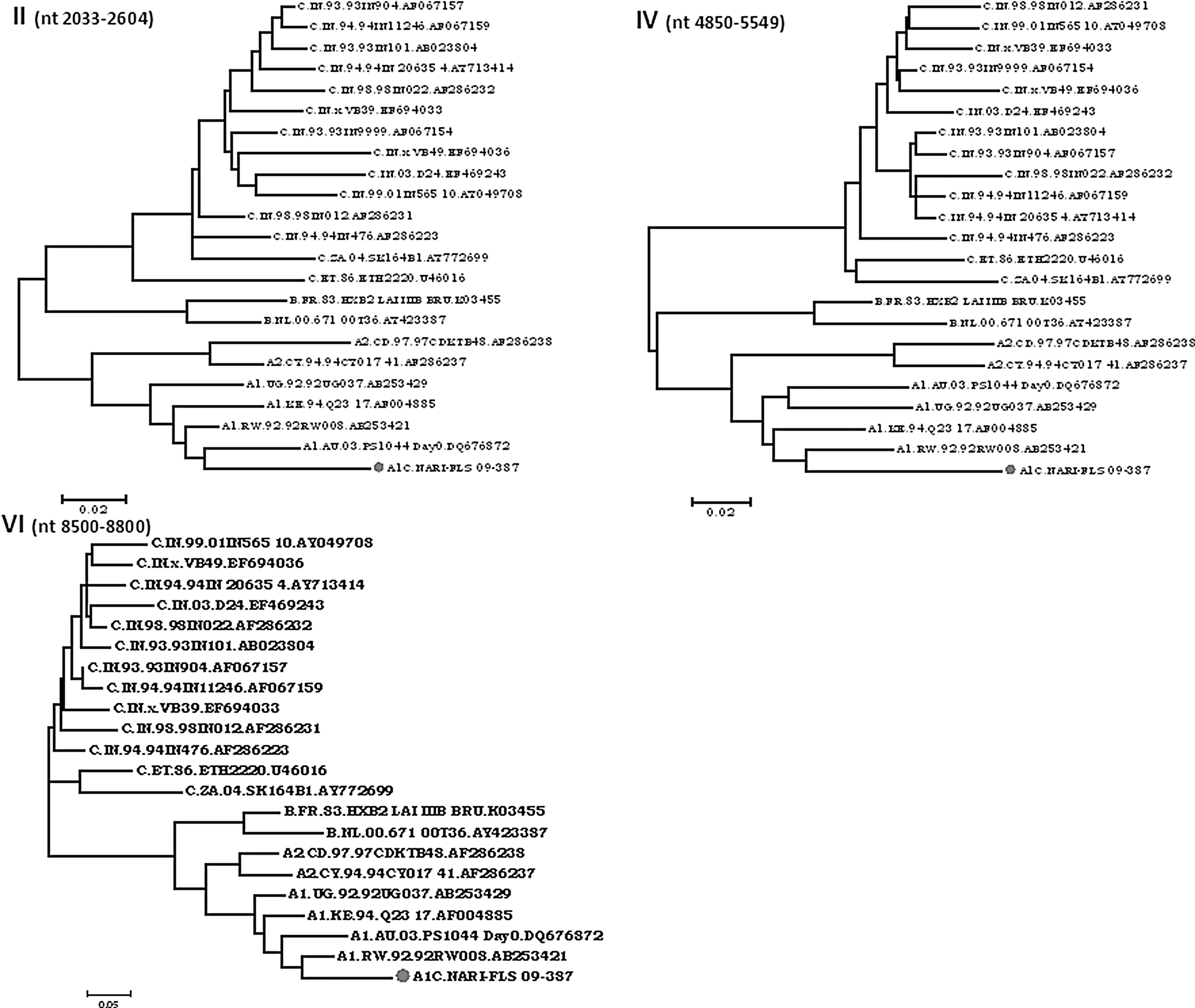
Phylogenetic analysis of segments (II, IV, and VI) showing subtype A1. Isolate NARI-FLS_09-387 clustered within subtype A1 radiation with Indian clade A1 sequences.
Phylogenetic analysis of segment III was further done with the breakpoints based on SimPlot results. Based on results obtained by SimPlot analysis segment III is composed of three blocks (A, B, and C) showing a small insertion of A1 (block B 4343–4519) within the clade C backbone. Block A (2604–4342) and block C (4520–4848) clustered within subtype C radiation with significant bootstrap values of 100 and 83, respectively (Fig. 7). Block B showed A1 in bootscan analysis, with a fall in A1 radiation with a bootstrap value of 63 with the isolate from Kenya but with a nonsignificant bootstrap value of 38 at the node of clade A1. BLAST search results indicated that block B had the highest similarity (97%) with subtype A1 sequences from Kenya.
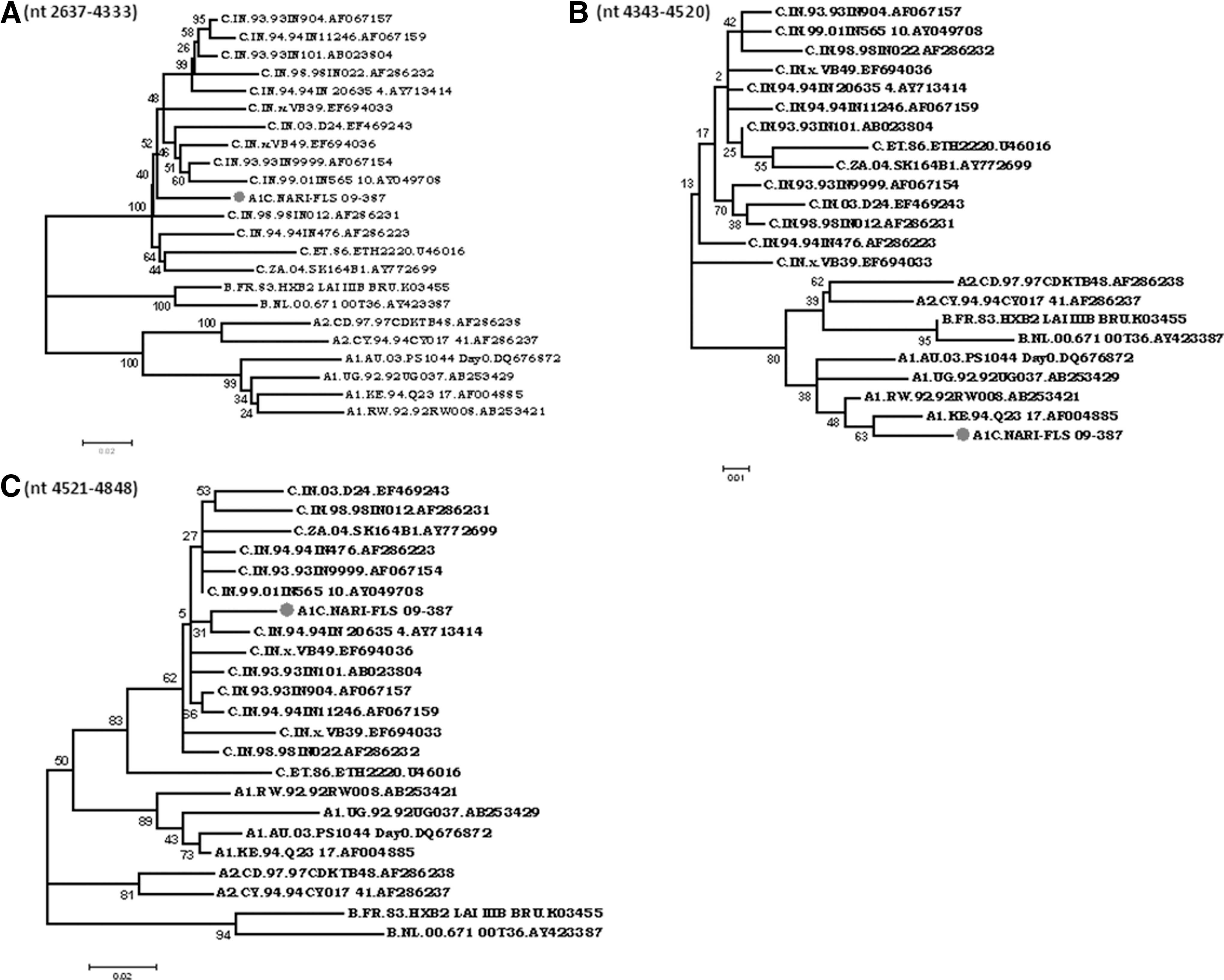
Phylogenetic analysis of blocks
Similarity with Kenyan isolates may be the result of the availability of very few HIV-1 subtype A sequences from India. Sliding window size has an important role in recombination identification. Zhang et al. 19 suggested that a window size of 300 was best suited for the identification of HIV-1 recombination. Discrepancies in the results by jpHMM@GOBICS and SimPlot may be due to the use of different window sizes. All the clones and DNA sequences clustered very closely (data not shown) and exhibited similar breakpoints as well as infection established from a single recombinant isolate.
The mosaic isolate NARI-FLS_09-387 shows recombination at four positions whereas Lole et al. 5 reported the A1/C mosaic virus with insertion of the clade A1 sequence only in the env-nef region (6320–9100 in the HXB2 coordinate) with the backbone of clade C. The other mosaic virus reported by Rodriguez et al. 6 showed recombination of C in the backbone of A1 at one position each in the gag (1134–1814) and pol (2623–4394) genes. Comparison with other mosaic viruses suggests gag (1800–2000 position) and pol (2600–2650) are the positions that favor the possibility of recombination.
Discussion
HIV-1 subtype A was reported in India as early as 1998. There is evidence available that subtype C has a fitness advantage over subtype A1 and it may be a possible cause for the relatively low prevalence of HIV-1 subtype A. 20 However, subtype A and C recombinants, described since 1998, have been reported from different parts of the country. The replication fitness and transmission potential of such strains need to be studied.
Since most of HIV-1 subtype A strains reported from India have been typed based on Heteroduplex Mobility Assay (HMA) or sequencing of a short genome fragment it is possible that they may not be pure HIV-1 subtype A but could be recombinants.
Recombination produces mosaic virus from parental strains and thus provides a shortcut to speed up viral evolution. All the recombinations may not necessarily provide a fitness benefit. However, rapidly increasing the proportion of CRFs globally indicates that the recombinant viruses may enjoy advantages over the parent strains. The relationship between recombination, mutation, and fitness is highly complex. Syncytium induction and multiple coreceptor usage are strongly associated with a greater decrease in CD4+ T cell count over time and a greater risk of disease progression and drug resistance. CTL escape mutants are major challenges to antiretroviral therapeutics and vaccine development. The phenotype of the newly defined CRF14_BG has been described as syncytium inducing, with usage of CXCR4, CCR5, CCR3, and CCR2b coreceptors. 21
In this study we characterized the full-length genome of the mosaic HIV-1 isolate NARI-FLS_09-387. The recombinant isolate shows insertion of subtype A1 at four positions in the backbone of subtype C. Clustering of the subtype C portion with Indian HIV-1 isolates suggests its Indian origin. There are very few partial and no full-length sequences of subtype A reported from India. This made the ancestral analysis of the subtype A1 portion difficult. This emphasizes the need to generate full-length genome sequence data and elaborate molecular characterization of all clade A1 isolates from India. In conclusion, this isolate is recombinant between subtype A1 and Indian clade C. This novel mosaic structure of the isolate is different from all other reported CRFs and URFs.
Sequence Data
The nucleotide sequences NARI-FLS_09-387 and its nine clones have been submitted to GenBank and the accession numbers are KC911635, KC911636, KC911637, KC911638, KC911639, KC911640, KC911641, KC911642, KC911643, KC911644.
Footnotes
Acknowledgments
We thank NARI clinics for providing us with clinical samples. This work was supported in part by a grant from the Department of Biotechnology (DBT), Government of India (BT/PR7054/Med/14/936/2006) to Ramesh Paranjape. We thank the Council of Scientific and Industrial Research (CSIR), Government of India for providing a senior research fellowship to Sudhanshu Pandey.
Author Disclosure Statement
No competing financial interests exist.