
Abstract
Genetic polymorphisms within the MHC encoding region have the strongest impact on HIV disease progression of any in the human genome and provide important clues to the mechanisms of HIV immune control. Few analyses have been undertaken of HLA alleles associated with rapid disease progression. HLA-B*07:02 is an HLA class I molecule that is prevalent in most populations worldwide and that has previously been consistently linked to accelerated disease progression in B-clade infection. This study investigates the observation that HLA-B*07:02 is not associated with a high viral setpoint in C-clade infection. We examine the hypothesis that this clade-specific difference in association with disease outcome may be related to distinct targeting of CD8+ T cell epitopes. We observed that C-clade-infected individuals with HLA-B*07:02 target a broader range of Gag epitopes, and to higher magnitudes, than do individuals infected with B-clade infection. In particular, a novel p17-Gag (Gag22-30, RPGGKKHYM) epitope is targeted in >50% of HLA-B*07:02-positive C-clade-infected individuals but clade-specific differences in this epitope result in nonimmunogenicity in B-clade infection. Only the C-clade p24-Gag “GL9” (Gag355-363, GPSHKARVL) epitope-specific CD8+ T cell response out of 16 studied was associated with a low viral setpoint. Although this epitope was also targeted in B-clade infection, the escape mutant S357S is present at higher frequency in B-clade infection than in C-clade infection (70% versus 43% in HLA-B*07:02-negative subjects). These data support earlier studies suggesting that increased breadth of the Gag-specific CD8+ T cell response may contribute to improved HIV immune control irrespective of the particular HLA molecules expressed.
Introduction
T
First, the Gag specificity of the CD8+ T cell response has been linked to immune control 7,10,11 exemplified by dominant Gag-specific responses restricted by protective HLA-B alleles, such as HLA-B*27 and HLA-B*57. 12 –15 Escape mutations from these responses that occur within the structurally conserved Gag protein typically result in reduced viral replicative capacity. 15 –19
Second, the HLA alleles associated with low viral load setpoints mostly carry the HLA-Bw4 motif, 20 which may also reduce viral load through interaction with KIR3DS1 and KIR3DL1 on natural killer cells. 21,22
Third, restrictive peptide-binding motifs, such as those found for the protective HLA-B*27 and HLA-B*57 alleles, 4,12,15,23 may reduce the number of self-reactive peptides bound during thymic selection, leaving a broader T cell receptor (TCR) repertoire available in the periphery to accommodate HIV viral sequence diversity. In contrast, alleles traditionally associated with “disease susceptibility,” including HLA-B*07 and HLA-B*35, bind a broader repertoire of self-peptides leaving a more narrow TCR repertoire in the periphery for recognition of viral sequence diversity. 24
Additional factors that may influence control of HIV via HLA expression include the interaction of HLA class I with leukocyte immunoglobulin-like receptors (LILRs) expressed on dendritic cells. This mechanism, leading to impaired dendritic cell function, has been proposed to contribute to the more rapid progression to HIV disease in people expressing certain HLA-B*35 subtypes. 25
We recently showed that HLA-B*35:01, an allele consistently associated with disease progression in B-clade infection, 3,26 is somewhat protective in C-clade infection. This outcome hinges on a specific CD8+ T cell response to a single Gag epitope only available in C-clade infection. 27 Similarly, we observed that HLA alleles belonging to the B*07 superfamily, including HLA-B*07:02, were associated with disease progression in B-clade infection, but not in C-clade infection. Consistent with these findings, recent studies of 3,622 B-clade-infected study subjects found that HLA-B*07:02 was associated with disease progression, 3,28 but this was not the case in a large cohort of 1,210 C-clade-infected individuals from Durban, South Africa. 6
We here test the hypothesis that, like HLA-B*35:01, subjects with HLA-B*07:02 impose a broader and more dominant Gag-specific CD8+ T cell response in C-clade infection leading to improved virologic outcomes, compared to a more narrow Gag-specific response in B-clade infection that is associated with disease progression.
Materials and Methods
Ethics statement
Ethics approval was given by University of KwaZulu-Natal Review Board and the Massachusetts General Hospital Review Board (Durban cohort), the Office of Human Research Administration, Harvard School of Public Health, and the Oxford Research Ethics Committee (Thames Valley and other cohorts). Study subjects from all cohorts gave written informed consent for their participation.
Study cohorts
We studied a total of 2,718 adults with chronic, antiretroviral therapy (ART)-naive HIV-1 infection, recruited from four cohorts as follows: (1) Durban, South Africa (C-clade, n=1,218), as previously described, 5,7,10,15 (2) Thames Valley cohort, UK (mixed clades, n=237), as previously described, 5,29 and (3) B-clade-infected cohorts from Kumamoto, Japan (B-clade, n=242) and (4) the United States (n=1,021), as previously described. 30 Viral loads were performed using the Roche Amplicor version 1.5 assay.
HLA typing and classification
HLA typing was undertaken from genomic DNA by sequence-based typing as previously described. 5 Locus-specific polymerase chain reactions (PCR) of exons 2 and 3 were amplified and sequenced.
Definition of HLA-B*07:02-restricted epitopes
To define a comprehensive list of HLA-B*07:02-restricted epitopes, we identified previously characterized epitopes from studies of predominantly B-clade-infected subjects (Los Alamos “A list”;
HLA-B*07:02-
Not listed in the Los Alamos “A” list database.
OLP, overlapping peptide.
Interferon (IFN)-γ ELISpot assays
IFN-γ ELISpot assays were undertaken using fresh or cryopreserved peripheral blood mononuclear cells (PBMCs). We screened for HIV-1-specific responses statistically associated (q<0.05) with the expression of HLA-B*07:02 by testing a total of 1,010 C-clade and 401 B-clade chronically infected subjects against a panel of 410 overlapping peptides (OLPs) spanning the entire HIV proteome, as previously described. 10,14 Significant associations were determined using Fisher's exact test and corrected for multiple comparisons using a q-value (FDR, false detection rate) approach as previously described. 7,14,32
We used B-clade and C-clade-specific optimal peptides to test for IFN-γ responses in HLA-B*07:02-positive B-clade-infected individuals recruited from the UK and Japan (n=58) and C-clade-infected individuals recruited from the UK (n=11).
Virus from all study subjects in the Japan cohort was sequenced to confirm clade of infection, and only those who were B-clade infected were included in the study. A response of 100 spot-forming cells (SFCs)/106 PBMCs was defined as significantly above the background response in control wells, which in most cases were zero.
Epitope fine mapping and HLA class I tetramer assay
We confirmed RM9-p17 (RPGGKKHYM), (Gag 22–30) as an optimal HLA-B*07:02-restricted epitope by stimulating PBMCs with truncated peptides in p-17-RM9 responder cells from ID: R045 and used peptide pulsed BCL lines partially HLA matching the responder p-17-RM9-specific 10 day expanded CTLs in an ICS assay. The corresponding p17-RM9 CTL line responses were validated using HLA-B*07:02 tetramers and controlled by a mismatched HLA tetramer. A pretitrated concentration of PE-conjugated tetramers 33 was used to stain p17-RM9-specific CTLs, incubated for 30 min and stained with pretitrated extracellular antibodies CD8-Pacific Blue (BD Pharmingen) and CD3-PacificOrange (Invitrogen). Dead cells were excluded by using Vivid Live/dead marker (Invitrogen). FACS data were analyzed using FlowJo version 8.8.6 (Treestar, USA).
Peptide-MHC-binding studies
HLA-peptide-binding studies were undertaken using a luminescent oxygen channeling immunoassay (LOCI) as previously described. 34 We tested binding for 16 HLA-B*07:02 epitopes as shown. Binding assays were performed in quadruplicate; the reported result is the mean of the four values obtained. Stability of binding (binding half-life) was performed as described previously. 35 Briefly, biotinylated HLA-I heavy chain, 125I-labeled β2-microglobulin (B2m), and peptide were allowed to fold into peptide-HLA-I complexes in streptavidin-coated scintillation microplates (Flashplate PLUS, Perkin Elmer, Boston, MA) for 24 h at 18°C. Excess of unlabeled B2m was added and dissociation was initiated by placing the microplate in a scintillation reader (TopCount NXT, Perkin Elmer, Boston, MA) operating at 37°C. The scintillation signal was monitored by continuous reading of the microplate for 24 h. Half-lives were calculated from dissociation curves using the exponential decay equation in Prism v.5.0a (GraphPad, San Diego, CA). Assays were performed in duplicate; the mean value of two experiments is reported.
Statistical analysis
Statistical analysis was undertaken using GraphPad Prism v.5.0a (GraphPad, San Diego, CA). Overlapping peptide responses and HLA expression were determined using Fisher's exact test and corrected for multiple comparisons using a q-value (false detection rate), as previously described. 7,32 Comparing responders and nonresponder subjects for peptide recognition and viral sequences was determined by Fisher's exact test. The Mann–Whitney U test was used to compare viral load setpoints for GL9-Gag in B-clade-infected and C-clade-infected individuals. Correlation between percent optimal peptide recognition and peptide binding IC50 values and peptide-binding half-lives (hours) was determined by Spearman rank correlations. Data presentation and statistical analysis were undertaken by GraphPad Prism v.6.0c.
Results
Dominant Gag-specific HLA-B*07:02-restricted CD8+ T cell responses in C-clade infection compared to B-clade infection
Previously we had shown no impact of HLA-B*07:02 on viral setpoint in a highly-powered study of >2,000 HIV-infected subjects in Southern Africa. 6,36 –38 In contrast, several studies have consistently shown a strong independent effect of HLA-B*07:02 on disease progression in B-clade infection, 3,26,28 but not in C-clade infection, 6,27 and therefore suggest a consistent impact of HIV clade on the association of HLA-B*07:02 with rapid HIV disease progression.
To determine whether the observed differential HLA-B*07:02-associated HIV disease outcomes in B-clade and C-clade-infected cohorts are related to clade-specific differences in CD8+ T cell responses, we tested a set of 16 clade-specific HLA-B*07:02-restricted optimal epitopes (Table 1). This panel comprised previously defined optimal epitopes that are detailed within the Los Alamos “A-list” 31 and an additional four epitopes identified by testing 1,010 C-clade-infected 10 and 401 B-clade-infected individuals 39 for recognition of 410 overlapping peptides spanning the C-clade and B-clade proteomes, respectively. Based on these screenings we suggest an additional four epitopes restricted by HLA-B*07:02 and not listed in the Los Alamos “A-list” (Table 1). The novel epitope, RPGGKKHYM (Gag 22-30) (p17-RM9), was optimized and unequivocally defined as restricted by HLA-B*07:02 (Fig. 1).
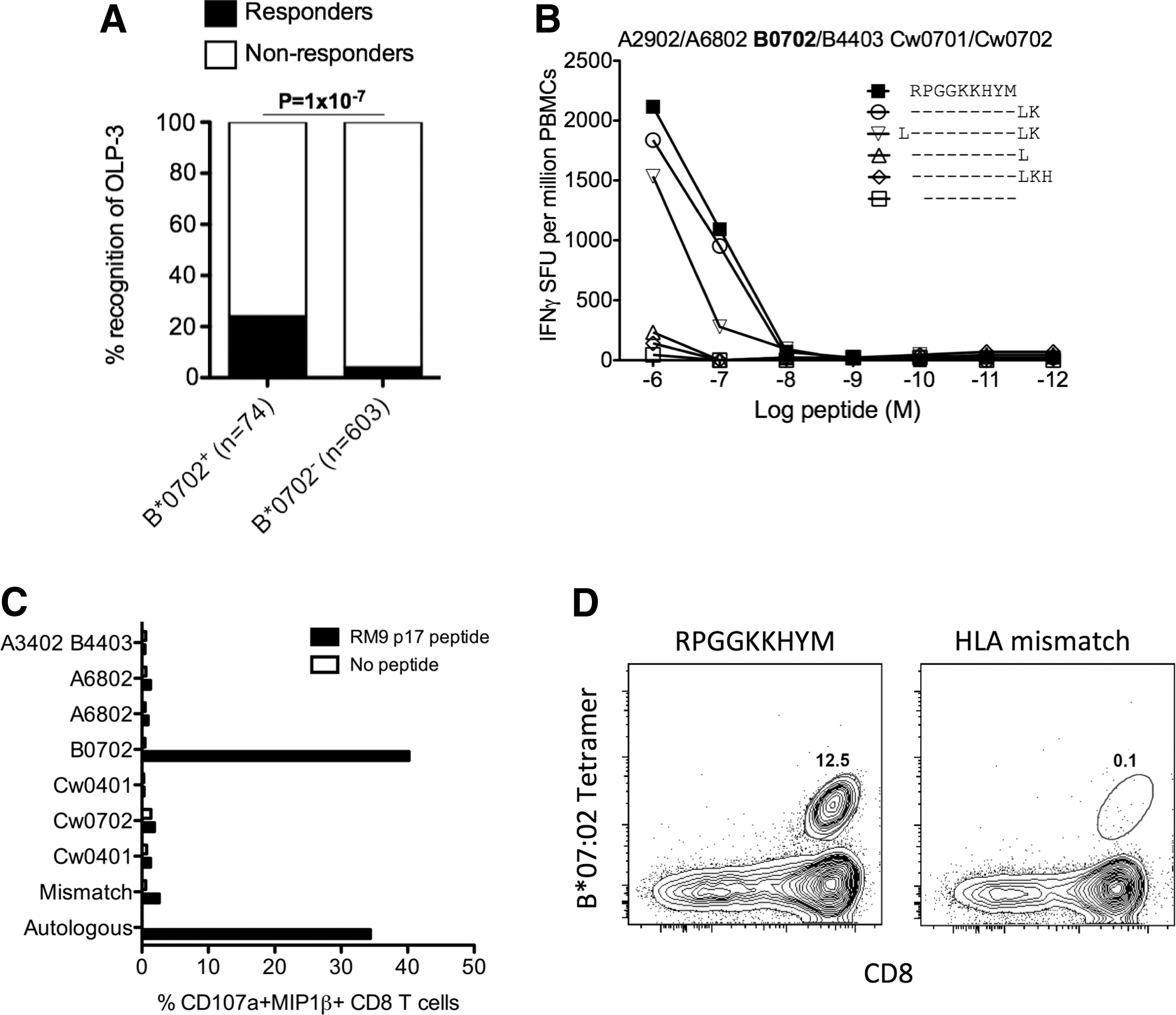
Identification and characterization of the p17-RM9 Gag epitope.
Reactivity to this comprehensive panel of clade-specific optimal epitopes was tested in HLA-B*07:02-positive subjects with B-clade infection (n=58) and in subjects with C-clade infection (n=11) using IFN-γ ELISpot assays (Fig. 2). We observed significantly more responses directed toward Gag for C-clade compared to B-clade-infected individuals (p=0.02). At the individual epitope level, the statistically significant clade-specific difference was the response to two Gag epitopes, p17-RM9 and p24-TL9, targeted more by C-clade-infected individuals compared to B-clade-infected individuals (p=4×10–5 and 1×10–3, respectively) (Fig. 2B), whereas Vif-HI10 was targeted only by B-clade-infected individuals, p=0.05 (Fig. 2D). Of note, no significant difference in cross-recognition of the p24-GL9 “3S” (C-clade) and “3G” (B-clade) version was observed for 6/11=55% and 17/58=29% in C-clade and B-clade-infected individuals, respectively (p=0.16) and confirmed by peptide titration in three C-clade and two B-clade-infected subjects (data not shown). Increased magnitudes of Gag and Nef-specific responses were detected for C-clade-infected compared to B-clade-infected individuals (p<0.05) (Fig. 2E) with increased breadth of Gag-specific epitope targeting for C-clade compared to B-clade-infected individuals (p<0.02) (Fig. 2F).
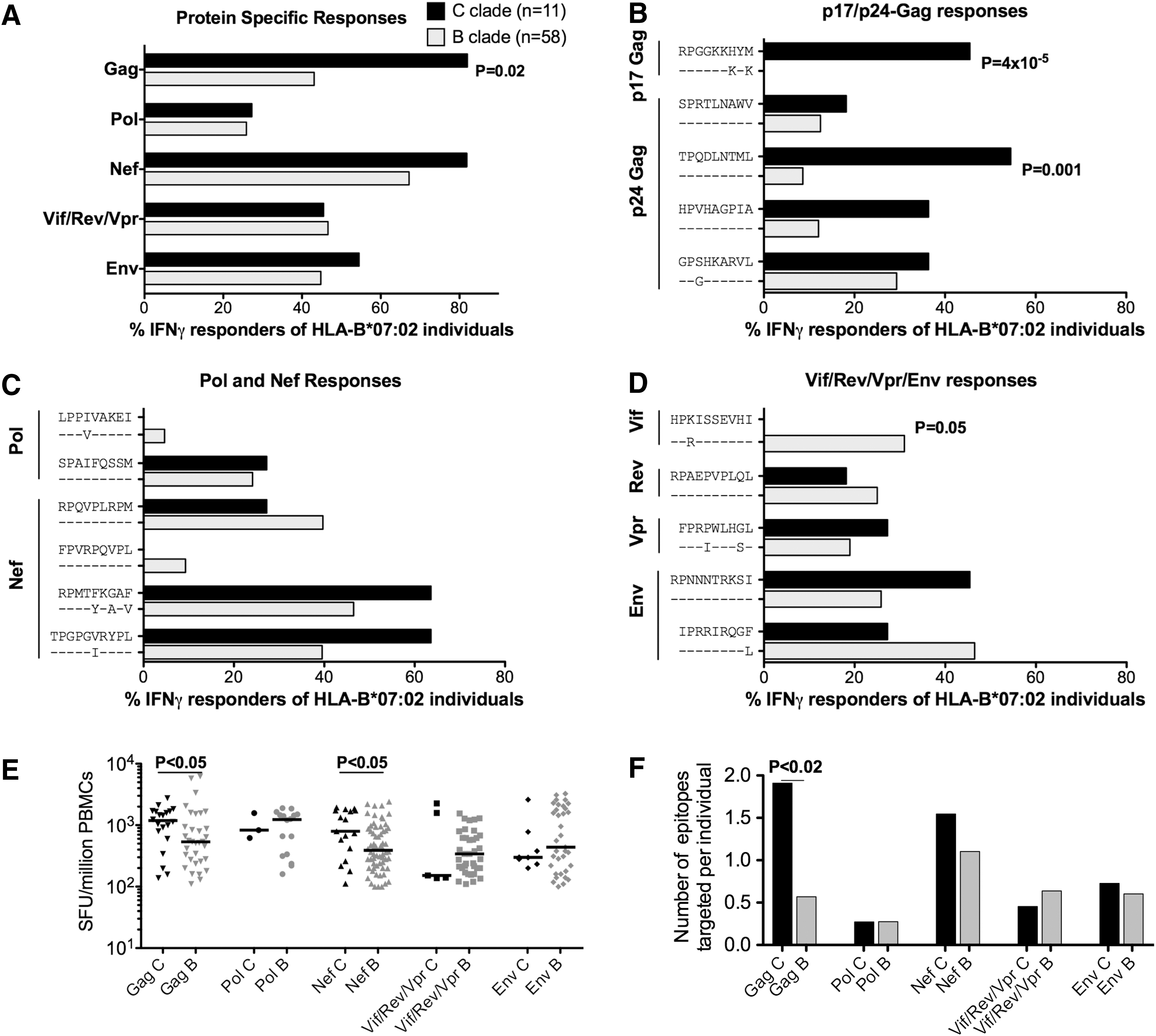
Percentage of HLA-B*07:02 subjects making IFN-γ ELISpot responses to 16 B-clade and C-clade-specific HLA-B*07:02-restricted optimal epitopes in B-clade and C-clade-infected subjects.
Clade-specific viral sequence differences determine epitope immunogenicity
The single HLA-B*07:02-restricted epitope not targeted in B-clade infection is the novel Gag epitope p17-RM9, which differs from the C-clade consensus sequence at two positions, H7K and M9K. The change M → K at the carboxy-terminal position (PC) does not fit the preferred peptide-binding motif of isoleucine/leucine at the C terminal residue in the F pocket of HLA-B*07:02. 23 Peptide-binding half-life (stability) studies were undertaken, revealing that only the C-clade version was stable in complex with HLA-B*07:02 (Fig. 3A), which was then confirmed by the determination of peptide-binding affinities to HLA-B*07:02 (RPGGKKHYM vs. RPGGKKKYK, K d=14 nM and >20,000 nM, respectively, Table 1). The nonimmunogenicity of p17-RM9 in B-clade infection was further confirmed by lack of cross-recognition from C-clade p17-RM9-specific CD8+ T cells against the nonbinding B-clade version of this epitope (Fig. 3B). A subset of clade B-infected subjects recognized the C-clade version of the p17-RM9 epitope, although still significantly less frequent than C-clade-infected individuals (4/58=7%, p=0.004) (data not shown). In addition, the single epitope, Vif-HI10, targeted significantly more in B-clade infection and nonimmunogenic in C-clade-infected individuals, had a 40-fold stronger binding affinity in the B-clade (K d=4 nM and 178 nM, in B-clade and C-clade, respectively) and was also less stable in complex with HLA-B*07:02 (half-life 4.3 h versus 2.3 h, respectively) (Table 1). Overall, we observed a weak correlation between peptide-binding affinity and binding half-life (stability) and the frequency of epitope recognition (r=0.34, p=0.06 and r=–0.4, p=0.03, respectively) (Fig. 3C and D) as previously observed for other HLA-B-restricted responses. 14
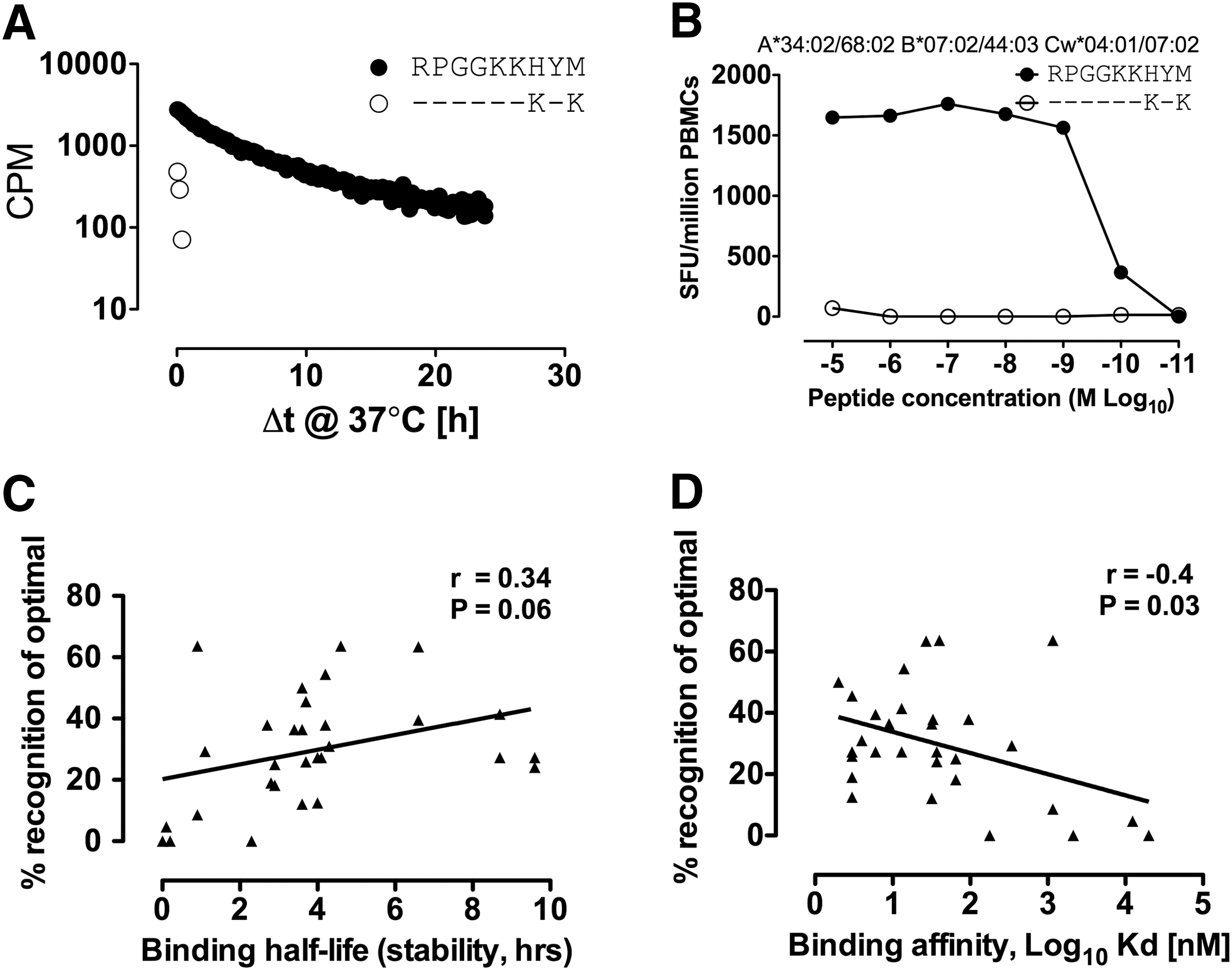
Clade-specific differences influence HLA-B*07:02 peptide binding and dictate epitope immunogenicity.
p24 Gag-GL9 response in C-clade infection is associated with lower viral setpoint
To assess the antiviral efficacy of each of the 16 HLA-B*07:02-restricted responses in C-clade and B-clade infection (Table 1), we compared the viral load setpoint of responding and nonresponding HLA-B*07:02-positive individuals and found that the single response associated with a significantly lower viral load setpoint was the C-clade version of the p24-GL9 (Gag 355–363, GPSHKARVL) specific response (p=0.001, Fig. 4A). In contrast, this response was not associated with any change in viral load setpoint in B-clade-infected individuals when tested against both the B-clade and C-clade versions of this peptide (data not shown).
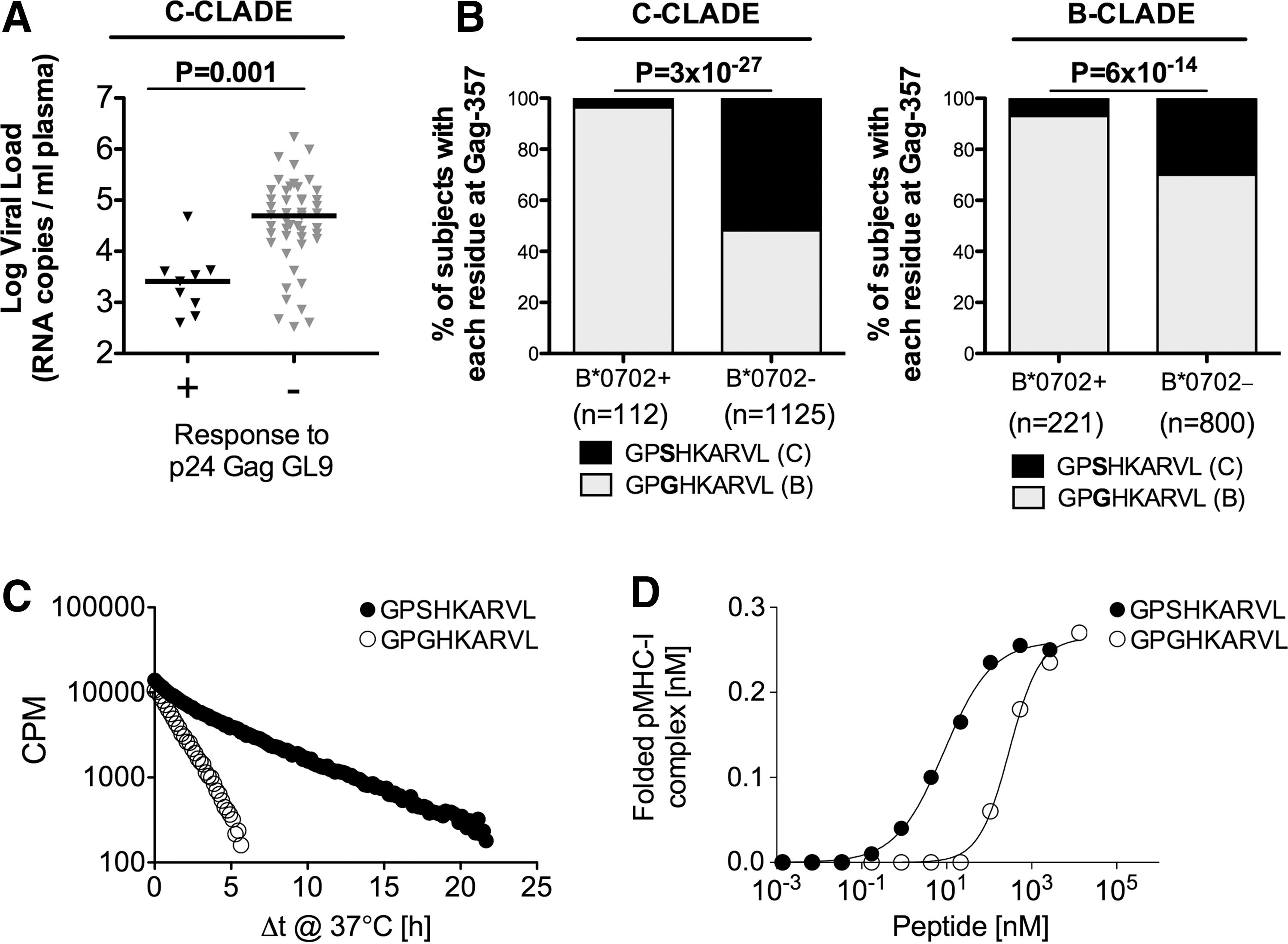
Peptide binding and stability influence antiviral activity and HIV selection pressure.
Approximately one-third (36%) of HLA-B*07:02-positive individuals show detectable responses to p24-GL9 in chronic C-clade infection, but this response drives strong selection pressure in acute infection 30 on the virus for the selection of the S357G escape mutant: 96% of HLA-B*07:02-positive subjects carry the GL9-357G mutation compared to 43% of HLA-B*07:02-negative subjects (p=3×10–27) (Fig. 4B). The GL9-357G mutation is also selected in HLA-B*07:02-positive subjects in B-clade infection, but 70% of B-clade sequences carry GL9-357G. In addition, the S357G mutation has a significant impact on the peptide-binding half-life and binding affinity (3.4 h vs. 1.1 h and K d== 9 nM vs. 342 nM, respectively) (Fig. 4C and D) (Table 1).
Taken together, these data show that clade-specific differences influence epitope immunogenicity and antiviral CD8+ T cell response efficacy. Alteration of the peptide-HLA-B*07:02 interaction ultimately results in improved Gag-specific epitope targeting in C-clade infection over B-clade infection at the population level.
Discussion
We consistently show here that HLA-B*07:02 is not inherently linked to HIV disease progression, but that individuals infected with a C-clade virus have improved immune control compared to individuals infected with a B-clade virus. Overall, individuals infected with a C-clade virus had a greater breadth and magnitude of HLA-B*07:02-restricted responses targeting Gag epitopes. At the individual epitope level, C-clade-infected individuals targeted two Gag epitopes significantly more often, one in p17-RM9 Gag and one in p24-TL9 Gag, whereas B-clade-infected individuals targeted the Vif-HI10 epitope significantly more frequently. Clade-specific differences in the p17-RM9 Gag epitope resulted in nonbinding and therefore nonimmunogenicity in B-clade infection.
A second observation was that, for the p24-GL9 Gag epitope, targeting frequencies did not differ significantly between B-clade and C-clade-infected subjects, but a response to this epitope was associated with lower viral load setpoints only in C-clade infection. This may be related to the fact that the consensus sequence in B-clade is predominantly the relatively poor binding variant and therefore elicits a different qualitative CD8+ T cell response compared to the S357 C-clade version of the epitope. In addition, the strong selection of the G357S mutation by HLA-B*07:02 expressing individuals in both C-clade and B-clade infection, but the higher frequency of consensus B-clade version of the p24-GL9 epitope (3G) in the HLA-B*07:02-negative B-clade-infected individuals compared to HLA-B*07:02-negative C-clade-infected individuals, may be a consequence of the higher frequency of HLA-B*07:02 expression in the B-clade (22%) than in the C-clade (9%)-infected cohorts studied here. 30 It seems that the S357G mutation is selected very early in acute infection and tends not to revert posttransmission, so this may be an example of a mutant that would accumulate over the course of the epidemic, especially rapidly in populations where HLA-B*07:02 is highly prevalent. 30
These data support the recent study on clade-specific differences in HLA-B*35:01 in control of HIV infection, which indicated that the protein specificity of the CD8+ T cell response makes an important contribution to viral setpoints and therefore to HLA associations with HIV disease outcome. 27 These HLA molecules are both HLA-Bw6 and therefore do not contribute to the HLA-B-KIR interactions that previous studies have shown make important contributions to immune control of HIV. 21,22 It is also striking that HLA-B*07:02 and HLA-B*35:01, which are the two HLA class I molecules that have been proposed to precipitate rapid HIV progression as a result of a more narrow TCR repertoire, 24 both are linked to a better immune control in C-clade infection along with other HLA-B alleles belonging to the B*07 superfamily. 6,27 These studies therefore are consistent with the hypothesis that increased targeting of Gag epitopes is associated with immune control.
In summary, this study examines a clade-specific difference in mechanisms of immune control mediated by HLA-B*07:02, in which B-clade is associated with rapid progression, while C-clade is not. The data presented indicate that clade-specific differences can alter the CD8+ T cell response, and suggest that these have an important impact on sustained control of HIV. 40 This suggests that immune control of viral replication can be achieved, irrespective of the restricting HLA molecule. 10,41 Ultimately, this provides hope that a vaccine focused on inducing responses targeting conserved regions of the proteome such as Gag will be effective, regardless of the HLA expression of the infected individual.
Footnotes
Acknowledgments
This work was supported by the Welcome Trust (P.G.) and the National Institutes of Health, grant R01 AI46995. H.N.K holds a grant from the Danish Agency for Science, Technology and Innovation #12-132295.
H.K. performed the study and wrote the paper. E.A. and M.K. performed experiments. A.S. generated tetramers. M.H. performed peptide HLA binding assays. P.M. analyzed data. C.B., B.W., T.N., and M.T. established and oversaw the HIV cohorts. S.B. supported and established tetramer and peptide binding data. P.G. provided supervision and financial support.
Author Disclosure Statement
No competing financial interests exist.