
Abstract
Innate immune responses to microbial pathogens are initiated following the binding of ligand to specific pattern recognition receptors. Each pattern recognition receptor, which includes members of the Toll-like receptor (TLR) family, is specific for a particular type of pathogen associated molecular pattern ensuring that the organism can respond rapidly to a wide range of pathogens including bacteria, viruses, and fungi. We studied the extent to which agonists to endosomal TLR could induce anti-HIV-1 activity in peripheral blood mononuclear cells (PBMCs). When agonists to TLR3, TLR7, TLR8 and TLR9 were added prior to infection with HIV-1, they significantly reduced infection of peripheral blood mononuclear cells. Interestingly, agonists to TLR8 and TLR9 were highly effective at blocking HIV replication even when added as late as 48 h or 72 h, respectively, after HIV-1 infection, indicating that the anti-viral effect was durable and long lasting. Analysis of the induction of anti-viral genes after agonist activation of TLR indicated that all of the agonists induced expression of the type I interferons and interferon stimulated genes, although to variable levels that depended on the agonist used. Interestingly, only the agonist to TLR9, ODN2395 DNA, induced expression of type II interferon and the anti-HIV proteins Apobec3G and SAMHD1. By blocking TLR activity using an inhibitor to the MyD88 adaptor protein, we demonstrated that, at least for TLR8 and TLR9, the anti-HIV activity was not entirely mediated by TLR activation, but likely by the activation of additional anti-viral sensors in HIV target cells. These findings suggest that agonists to the endosomal TLR function to induce expression of anti-HIV molecules by both TLR-mediated and non-TLR-mediated mechanisms. Moreover, the non-TLR-mediated mechanisms induced by these agonists could potentially be exploited to block HIV-1 replication in recently HIV-exposed individuals.
Introduction
I
Several of the TLR reside within endosomes and recognize microbial nucleic acids during the initial infectivity event. 2 Viruses whose genome is composed of single-stranded RNA are recognized by TLR7 and TLR8. Activation of these TLR following ligand binding induces intracellular signaling pathways mediated via the cytoplasmic adaptor protein MyD88 that induces the cell to produce interferon (IFN)-α, along with inflammatory cytokines and chemokines designed to thwart the infection. TLR9 recognizes unmethylated microbial DNA containing the sequence CpG, and activation of this receptor following ligand binding also induces the recruitment of MyD88 and the production of proinflammatory cytokines. 4 TLR3 recognizes double-stranded RNA molecules, and in contrast to the other TLR that signal through MyD88, activation of this TLR results in the phosphorylation and activation of interferon regulatory factor 3 (IRF3). Once activated, IRF3 forms a complex with the cyclic AMP-response element binding protein (CREB) binding protein (CREBBP), translocates to the nucleus, and induces transcription of the type I interferons, IFN-α and IFN-β, along with interferon regulatory factor 7 (IRF7) that further increases type I interferon production.
Agonists to several of the TLRs have been studied for their ability to inhibit or moderate viral infections in animal and in cell culture models. Sariol et al. studied the protective effects of TLR3 and TLR7/8 agonists administered to rhesus macaques following dengue virus infection. 5 Macaques were exposed to dengue virus, followed by the administration of TLR3 and TLR7/8 agonists subcutaneously in emulsified form at 2 days and 7 days postinfection. In animals that received TLR agonists, viral replication was reduced, proinflammatory reactions were enhanced, and antidengue antibody titers were increased. These effects were attributed in part to the activation of myeloid dendritic cells, as increased serum levels of CXCL-10 and interleukin (IL)-1Rα were detected in animals administered TLR agonists. In contrast, animals that were exposed to dengue in the absence of TLR agonists experienced acute illness. 5
Ashkar et al. studied the role of TLR3, TLR4, and TLR9 activation in a murine model of herpes simplex virus-type 2 (HSV-2) infection. 6 Mucosal administration of the TLR9 agonist CpG oligodeoxynucleotide (ODN) induced an antiviral state that protected mice against intravaginal challenge with HSV-2, although this was accompanied by local inflammation and splenomegaly. In contrast, vaginal instillation of a TLR3 ligand also induced protection against HSV-2 challenge, but did so in the absence of local inflammation and splenomegaly. 6
Johnson et al. studied the role of TLR activation in immunization and viral challenge in a murine model of respiratory syncytial virus (RSV). In this study, TLR7/8 or TLR9 agonists were administered to mice concurrently with RSV immunization followed by challenge with RSV. When CpG, a TLR9 agonist, was administered together with an RSV immunogen, mice were found to have reduced disease severity, reduced viral titers in the lung, and lower levels of inflammatory cytokines following RSV challenge. TLR7/8 agonists, in contrast, had no beneficial effect. However, if either the TLR7/8 or TLR9 agonists were administered at the time of RSV infection, mice showed increased clinical symptoms and pulmonary inflammation. 7 These authors concluded that the immunostimulatory properties of TLR agonists enhanced disease severity when used during RSV infection rather than when used during immunization, suggesting a beneficial effect of TLR activation on enhanced immune responses.
The use of TLR ligands to moderate or inhibit HIV-1 infection has been studied both in vitro and in vivo. Victoria et al. 8 reported that the TLR2 agonists zymosan and Pam3Cysk4 added to human macrophages prior to infection with HIV-1 markedly reduced levels of HIV-1 replication. 8 Analyses of cytokine production by macrophages before and after TLR2 activation indicated that IL-10 and the β-chemokines CCL3, CCL4, and CCL5 were in part responsible for the antiviral effects. 8 Zhou et al. used poly(I:C), a TLR3 agonist, to demonstrate induction of anti-HIV innate immune factors in macrophages at levels that successfully inhibited HIV-1 infection and replication in these cells. 9 Interestingly, bacterial-derived lipopolysaccharide, a classical ligand for TLR4, was shown by Franchin et al. to inhibit macrophage infection by HIV-1 by altering the recycling of CCR5, resulting in the down-regulation of expression of this chemokine coreceptor. 10 We previously reported that gardiquimod, a TLR7 agonist, effectively inhibited HIV-1 replication in activated lymphocytes and macrophages by two distinct mechanisms. 11 As a TLR7 agonist, gardiquimod induced high levels of IFN-α, a cytokine that expresses potent antiviral activity. In addition, gardiquimod directly inhibited HIV-1 reverse transcriptase by functioning as a nucleoside reverse transcriptase inhibitor. 11 These findings indicate that TLR agonists may be capable of inhibiting HIV-1 infection and/or replication not only by direct activation of specific TLR, but also by other non-TLR-mediated mechanisms.
These TLR-mediated anti-HIV effects were not observed, however, in studies with a transgenic murine model as reported by Bafica et al. 12 Their transgenic murine model expressed the entire HIV-1 genome, including the long-terminal repeat regions. They demonstrated that several different TLR ligands induced HIV-1 gene expression in splenocytes and purified antigen-presenting cells isolated from these mice and cultured in vitro. When immune cells were rendered tolerant to TLR2, 4, or 9 activation by prolonged exposure to agonists to these TLR in vitro, they observed increased p24 production, indicating a reactivation of viral replication in the presence of TLR agonists. 12 Moreover, they observed similar effects when mice were exposed to TLR4 ligands in vivo. However, the clinical significance of these studies can be questioned by the fact that these mice do not express viral entry receptors and thus lack normal mechanisms for viral spread. A related study by Equils et al. 13 using a similar HIV transgenic mouse strain demonstrated that coactivation of TLR by bacterial ligands can activate HIV-1 transcription and lead to enhanced viral protein production. 13 These findings, and those of Bafica et al., 12 are interesting because of the exposure to gut bacteria in patients following bacterial translocation. Agrawal and Martin 14 postulated that the TLR9 agonist used in these studies, CpG, was responsible for the induction of HIV-1 transcription in these transgenic murine models. They noted that a clinical trial of GEM91, an antisense oligodeoxynucleotide similar to CpG, resulted in increased viral loads in study participants. 14 They cautioned that TLR9-mediated immune responses to the GEM91 were likely activating HIV-1-infected cells resulting in enhanced viral transcription. In a macaque model of vaginal simian immunodeficiency virus (SIV) transmission, Wang et al. showed that the mucosal application of TLR9 and TLR7 agonists resulted in an increase in the transmission rates of SIV and higher plasma viral loads. 15 They reasoned that the induction of type I interferons IFN-α and IFN-β led to a massive influx of mononuclear cells to the vaginal mucosa, which served to enhance viral infection and transmission. Thus, it is important to evaluate findings from in vitro and in vivo studies with a clear understanding of the interaction among cell types present in the population studied, and in light of the fact that in vivo studies include effects from the cellular influx of immune cells, whereas in vitro studies exclude this variable.
Based on the apparent conflicting findings with respect to TLR activation and HIV-1 replication, we determined the extent to which endosomal TLR activation could alter HIV-1 replication in populations of activated peripheral blood mononuclear cells (PBMCs). We postulated that these mechanisms are likely to involve multiple intracellular signaling pathways leading to cellular activation and innate immune factor secretion. Some of the factors that express antiviral activity include the interferon-induced proteins such as myxovirus resistance gene (MxA), 16 2′-5′-oligoadenylate synthase (OAS), 17 double-stranded RNA-dependent protein kinase R (PKR), 18,19 ribonuclease L (RNAL), 17 and its endogenously expressed and HIV-induced suppressor protein termed ribonuclease L inhibitor (RLI). 20 Two additional HIV-specific restriction factors that impart potent HIV inhibitory activity include apolipoprotein B mRNA-editing catalytic 3G (Apobec3G) 21 and SAM-domain and HD-domain containing protein 1 (SAMHD1). 22 All of these proteins have been shown to be critically important for the antiviral innate immune response. Our particular interest focused on whether different endosomal TLR expressed varying patterns of interferon-stimulated gene expression and anti-HIV-1 activity in response to ligand binding. This could result from differences in cellular expression, as monocytes, macrophages, and myeloid-derived dendritic cells primarily express TLR3 and TLR8, whereas plasmacytoid dendritic cells, macrophages, B cells, and CD4+ and CD8+ T lymphocytes express TLR7 and TLR9. 23 –28
Our current studies sought to determine antiviral gene expression and HIV inhibition induced by agonists to TLR3, TLR7, TLR8, and TLR9, all of which reside in endosomes and are primary PRR that recognize microbial nucleic acids. The addition of an MyD88 inhibitor prior to the addition of agonists to TLR8 and TLR9 blocked induction of IFN-α but did not reverse the anti-HIV effect. This finding indicates that the antiviral effect was likely mediated by several different mechanisms. In support of this, we found that the addition of a broad-acting tyrosine kinase inhibitor, sunitinib malate, reversed the anti-HIV effect in cells treated with agonists to TLR7, TLR8, and TLR9. Sunitinib malate has been reported to block the activity of RNase L, an enzyme induced by interferon that destroys cellular and viral RNA, 17 as well as PKR (protein kinase RNA-activated), an enzyme induced by interferon and activated by viral RNA. 29 These findings suggest that agonists to endosomal TLR likely mediate anti-HIV effects via two different mechanisms. The first mechanism involves the classical activation of TLR with subsequent induction of type I, and in some cases type II, interferons. The second mechanism most likely involves the activation by these agonists of antiviral proteins and alternative PRRs in the cytosol. These studies suggest that the utility of these agonists as anti-HIV-1 compounds extends beyond their function as classical TLR activators, to include a potential role in stimulating additional, and perhaps more potent, antiviral mechanisms in a far larger and more diverse population of HIV-1 target cells.
Materials and Methods
Reagents
Poly(I:C) (tlrl-picw, Invivogen, San Diego, CA) was used as a double-stranded RNA agonist for TLR3, gardiquimod (tlrl-gdqs), an imidazoquinoline analog, and CL264 (tlrl-c264e), an adenosine analog, were used as TLR7 agonists, ssPolyU RNA (tlrl-sspu) was used as a single-stranded RNA agonist for TLR8, and ODN2395 (tlrl-2395) was used as an unmethylated CpG DNA agonist for TLR9. All TLR agonists were reconstituted in sterile, endotoxin-free water. All agonists, with the exception of ssPolyU RNA, were added directly to cells. The ssPolyU RNA agonist was delivered via nanoparticles prepared with INTERFERin (Genesee Scientific, San Diego, CA) as described. 30 Briefly, the appropriate amount of ssPolyU RNA was mixed with 4 μl of INTERFERin in 100 μl of serum-free RPMI and vortexed for 15 s followed by a 15-min incubation at room temperature. The nanoparticle suspension was added directly to PBMCs.
The MyD88 inhibitory peptide (tlrl-pimyd, Invivogen) was used as an inhibitor of TLR8 and TLR9 activation at a final concentration of 10 μg/ml. Sunitinib malate (1611-25, Biovision, Milpitas, CA) was used as a putative inhibitor for PKR and RNase L. Sunitinib malate is a small-molecule inhibitor of multiple receptor tyrosine kinases, including vascular endothelial growth factor receptors, platelet-derived growth factor receptors, CSF-1 receptor kinases, RNase L, PKR, and the KIT receptor. Sunitinib malate was dissolved in dimethyl sulfoxide (DMSO) at a concentration of 5 mg/ml and used at a final concentration of 5 μg/ml.
Peripheral blood mononuclear cell (PBMC) cultures
Peripheral blood was obtained from healthy donors over the age of 18 who were donating platelets for patient use at the Dartmouth-Hitchcock Medical Center (Lebanon, NH). Leukocytes trapped in a filter during plateletpheresis and normally discarded were used as a source of PBMCs. All donors were screened for blood-borne pathogens prior to their platelet donation. No information about the donors other than their age and gender was obtained. The mononuclear cell fraction was isolated by Ficoll-Hypaque (Amersham, Piscataway, NJ), and PBMCs were isolated and cultured as previously described. 11 After the final wash in RPMI 1640 (Sigma-Aldrich, St. Louis, MO), PBMCs were resuspended in RPMI 1640 supplemented with 10% fetal bovine serum, 2 mM glutamine, 50 units/ml penicillin, and 50 μg/ml streptomycin (complete RPMI) and activated with 2 μg/ml phytohemagglutinin (PHA-P, Invitrogen, Carlsbad, CA) for 48 h at 37°C in a humidified atmosphere. After activation, PBMCs were washed three times in RPMI 1640 to remove the PHA-P and resuspended in complete RPMI at 2×106 cells/ml for all subsequent studies. Data presented in each figure represent the average values from triplicate wells from a single representative donor. All experiments, however, were repeated for at least three different donors.
Infection of PBMCs with HIV-1
In those experiments to assess the effects of TLR activation on HIV-1 replication, PHA-activated PBMCs were infected with 20 TCID50/ml of HIV-1Ba-L (R5 tropic strain) for 1 h, followed by three washes in complete RPMI to remove any unincorporated virus. The PBMCs were resuspended after the final wash in complete RPMI containing 10 units/ml of IL-2 and cultured in triplicate wells of a six-well plate prior to the addition of TLR agonists.
TLR activation using specific agonists
Agonists to TLR3 [20 μg/ml poly(I:C)], TLR7 (1 μg/ml CL264 or gardiquimod), TLR8 (0.156 μg/ml ssPolyU), and TLR9 (5 μg/ml ODN2395) were added to PHA-activated PBMCs to assess the effects on HIV-1 replication and to measure induction of antiviral genes. In studies that measured the effects of these agonists on HIV-1 replication, PHA-activated PBMCs were first infected with HIV-1 as described above, and then incubated with specific agonists for 7 days. To assess the time dependence of TLR activation on HIV-1 replication, specific agonists were added to PHA-activated PBMCs beginning 24 h prior to, or up to 72 h after, infection with HIV-1. HIV-1 p24 levels in culture supernatant were quantified by ELISA (Dupont, Wilmington, DE) on day 7 post-HIV infection.
Induction of the interferon and interferon-stimulated genes by TLR agonists
For studies of gene induction following TLR activation, PHA-activated PBMCs (2×106/well in triplicate wells) were incubated with media alone, or with 1 μg/ml gardiquimod, 0.156 μg/ml ssPolyU, or 5 μg/ml ODN2395. Cells were removed from triplicate wells at 6 h and 24 h posttreatment, centrifuged to form a cell pellet, and RNA was isolated from the cell pellet using the RNeasy Plus Mini Kit (Qiagen, Valencia, CA). To remove contaminating genomic DNA, an on-column DNase digestion was performed according to the manufacturer's specifications (Qiagen). Two micrograms of total RNA from each time point was reverse transcribed to cDNA using random hexamer primers in a 50 μl reaction volume as described. 30 Duplicate samples without added reverse transcriptase were run to verify the absence of genomic DNA contamination. Ten microliters of the cDNA reaction was used for real-time PCR measurements of mRNA levels for type I (IFN-α and IFN-β) and type II (IFN-γ) interferons, while 2.5 μl of the cDNA reaction was used for real-time PCR measurement of the interferon-stimulated genes MxA, 2′-5′-oligoadenylate synthase (OAS), double-stranded RNA-dependent PKR, RNAL, RLI, the innate immune factors apolipoprotein mRNA-editing catalytic 3G (Apobec3G), and SAMHD1. Data from each condition were normalized to expression levels of ribosomal protein L13A (RPL13A). Subsequently, the level of each transcript was expressed relative to that measured in cells prior to the addition of agonist (time 0). Data were analyzed using the ΔΔC t method. The forward and reverse primer sequences for these genes are listed in Table 1.
IFN-α, interferon-alpha; IFN-β, interferon-beta; IFN-γ, interferon gamma; MxA, myxovirus resistance gene A; OAS, 2′-5′-oligoadenylate synthase; PKR, double-stranded RNA-dependent protein kinase R; RNAL, RNase L; RLI, RNase L inhibitor; Apobec3G, apolipoprotein mRNA-editing catalytic 3G; SAMHD1, SAM domain and HD domain-containing protein 1; RPL13A, ribosomal protein L13A.
Inhibiting signaling pathways mediated by the myeloid differentiation primary response gene 88 (MyD88) protein
To determine the correlation between interferon gene expression and HIV-1 inhibition, we blocked the downstream signaling events from the universal adaptor protein MyD88 using the MyD88 inhibitory peptide. Activation of TLR7, TLR8, and TLR9 induces a signaling cascade starting with MyD88 that leads to interferon production. For studies to measure the effects on interferon gene expression, PHA-activated PBMCs were treated for 4 h with 10 μg/ml of the MyD88 inhibitory peptide or its control peptide. Following the 4-h incubation, cells from the control peptide- or MyD88 inhibitory peptide-treated groups were divided into three groups and incubated either with media alone, or with 0.156 μg/ml ssPolyU (TLR8 agonist), or 5 μg/ml ODN2395 (TLR9 agonist). Cell pellets were collected from all cultures 6 h after the addition of agonists for extraction of total cellular RNA as described above. Transcript levels for IFN-α, IFN-β, and IFN-γ in MyD88 inhibitor-treated cells were expressed relative to those measured in cells treated with the control peptide.
For studies to measure the effects of MyD88 inhibition on HIV-1 infection, PHA-activated PBMCs were first infected with 20 TCID50 HIV-1BaL for 1 h, washed, and incubated with either the MyD88 inhibitor or the control peptide for 4 h, followed by the addition of agonists to TLR8 or TLR9. Supernatant was collected on day 7 and p24 levels were quantified by ELISA.
Inhibition of putative antiviral compounds that function as tyrosine kinases
Sunitinib malate binds to the kinase domains of several enzymes that have been shown to have anti-HIV activity, including RNase L and PKR, and can inhibit the antiviral activity of these proteins with an IC50 of 1.4 and 0.3 μM, respectively. 18 To determine whether the tyrosine kinase inhibitor sunitinib malate could reverse the anti-HIV effect of TLR agonists, PBMCs were infected with 20 TCID50 HIV-1Ba-L for 1 h, washed, and treated with 5 μg/ml (10 μM) sunitinib malate or an equal volume of DMSO for 4 h, followed by the addition of either media, 1 μg/ml gardiquimod, 0.156 μg/ml ssPolyU, or 5 μg/ml ODN2395. Supernatant from PBMC cultures was collected on day 7 and p24 levels were quantified by ELISA.
Cell metabolic activity
Metabolic activity of TLR agonist-treated cells was measured using the CellTiter 96 AQueous One Solution Cell Proliferation Assay (Promega, Madison, WI). PHA-activated PBMCs (2×105 cells/well) were incubated with media alone, 1 μg/ml gardiquimod, 0.156 μg/ml ssPolyU, or 5 μg/ml ODN2395. After 7 days, 20 μl of CellTiter reagent was added to each well and the cells were incubated overnight at 37°C. Metabolic activity was determined by a colorimetric change of the substrate at 490 nm as measured by a spectrophotometer.
Statistical analysis
Analysis of datasets comparing two groups was performed by paired Students' t-test. Data were considered statistically significant at p≤0.05 and specific p values are noted in each figure legend. In all figures, data are represented as the mean±standard deviation of the mean (mean±SD) of triplicate samples from either two or three different blood cell donors.
Results
TLR activation inhibits HIV-1 replication in PBMCs
Agonists to TLR3 [poly(I:C), 20 μg/ml], TLR7 (CL264, 1 μg/ml), TLR8 (ssPolyU RNA, 0.156 μg/ml), and TLR9 (ODN2395 DNA, 5 μg/ml) were added to cultures of PHA-activated PBMCs either 1 day before, immediately after, or 24 or 48 h after infection with HIV-1. Each agonist inhibited HIV-1 replication when the cells were treated 24 h prior to infection (Fig. 1) as determined by p24 levels on day 7 postinfection. However, each agonist displayed differences in its ability to inhibit HIV infection when added after HIV-1 infection. For example, the TLR3 agonist poly(I:C) suppressed HIV-1 infection when added the day before or immediately after infection, but was ineffective when added 24 h or 48 h after infection. In contrast, the TLR7 agonist CL264 was effective at inhibiting HIV-1 replication only when added the day before infection. The TLR8 agonist ssPolyU RNA inhibited HIV infection when added at any of the time points up to 72 h postinfection. The TLR9 agonist ODN2395 DNA was the most efficacious as it could be added 72 h after infection and still inhibit HIV-1 replication. Viability and metabolic activity of TLR agonist-treated and HIV-infected PBMCs were unchanged compared to untreated controls that were either infected or not infected with HIV-1 (data not shown).
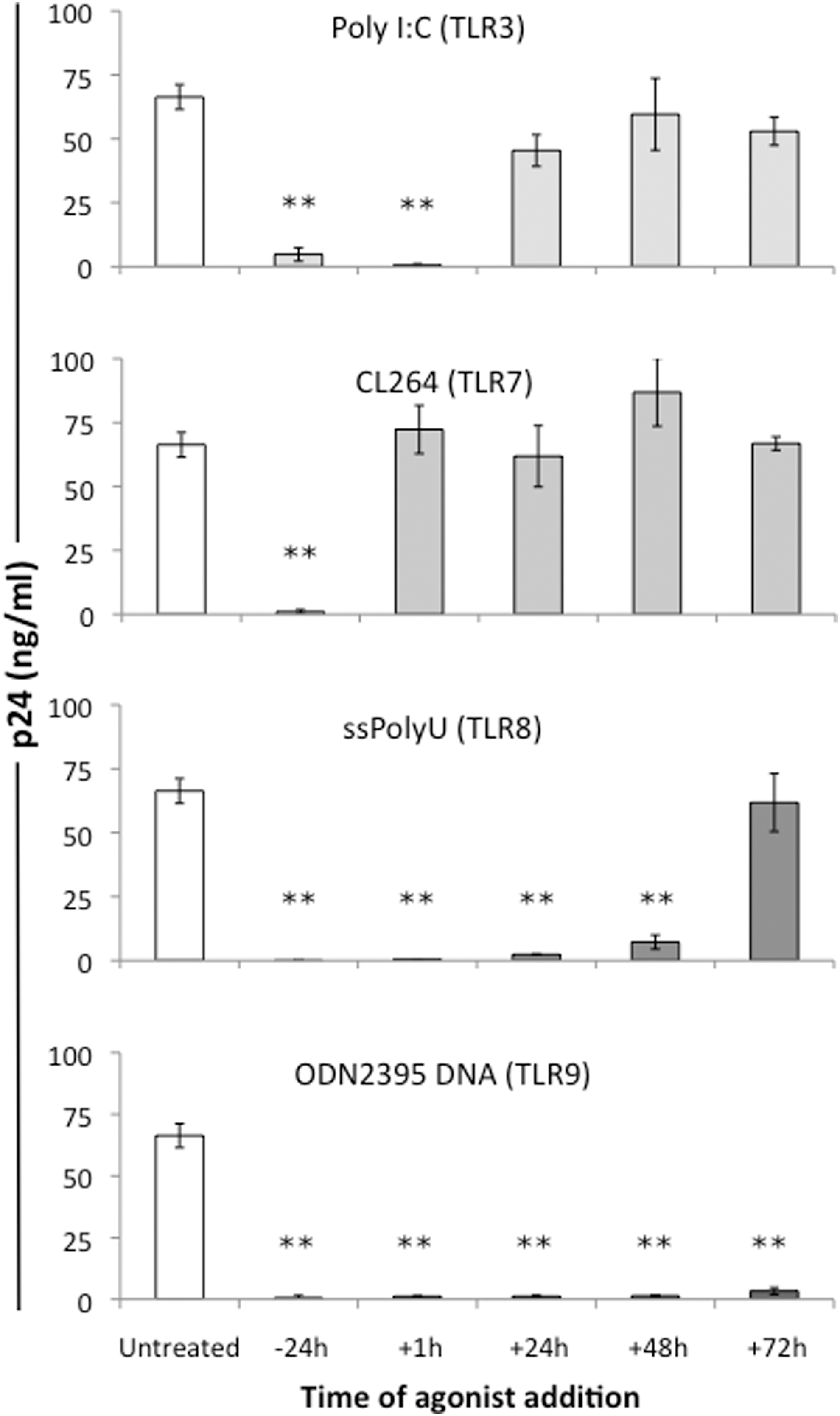
Toll-like receptor (TLR) agonists inhibit HIV-1 replication in peripheral blood mononuclear cells (PBMCs). Phytohemagglutinin (PHA)-activated PBMCs were treated once with agonists to TLR 3 [poly(I:C)], TLR7 (CL264), TLR8 (ssPolyU RNA), or TLR9 (ODN2395 DNA) either 24 h before, immediately after, or 24, 48, or 72 h post-HIV infection. p24 levels in culture supernatant were measured by ELISA on day 7 postinfection. All experiments were performed in triplicate using cells from three different donors. **p≤0.005.
Agonists to TLR7, TLR8, and TLR9 induce expression of antiviral genes in PBMCs
To determine the potential mechanism of TLR-induced HIV-1 inhibition, expression levels of antiviral genes were measured in PHA-activated PBMCs treated for 6 h with agonists for TLR7, TLR8, and TLR9. Treatment of PBMCs with gardiquimod (TLR7 agonist) resulted in a 10- to 30-fold increase in levels of IFN-α, and the interferon-stimulated genes (ISGs) MxA, OAS, and PKR (Fig. 2, upper panel). Stimulation of PBMCs with ssPolyU RNA (TLR8 agonist) resulted in a 100-fold increase in mRNA levels of the antiviral genes IFN-α and IFN-β and a 5- to 10-fold increase in MxA, OAS, PKR, and RNase L (Fig. 2, middle panel). Stimulation of PBMCs with ODN2395 (TLR9 agonist) resulted in a 100-fold induction of IFN-α and IFN-β expression and a 10-fold increase in the levels of MxA, OAS, and PKR when compared to untreated controls (Fig. 2, lower panel). Data from each of the agonist-treated groups were normalized to expression levels of ribosomal protein L13A (RPL13A), and the level of each transcript was expressed relative to that measured in cells prior to the addition of agonist, and expression levels arbitrarily set to “1.”

Activation of TLR7, TLR8, and TLR9 induces type I interferon and interferon-stimulated gene expression in PBMCs. PHA-activated PBMCs were cultured in triplicate with media alone or with 1 μg/ml gardiquimod (TLR7 agonist, upper panel), 0.156 μg/ml ssPolyU RNA (TLR8 agonist, middle panel), or 5 μg/ml ODN2395 DNA (TLR9 agonist, lower panel). Cells were harvested after 6 h, and RNA was isolated for real-time PCR analysis of the type I interferon (IFN-α, IFN-β) and interferon-stimulated gene (ISG) expression. Transcript levels for these genes from the untreated controls were arbitrarily set to “1” and are denoted by a dotted line. Data are plotted on a logarithmic scale. All experiments were performed in triplicate using cells from three different donors. PCR experiments had six replicates per condition. **p<0.005.
Type II interferon (IFN-γ) is induced by the TLR9 agonist ODN2395
We determined the extent to which TLR activation induces a type II interferon response by measuring the induction of IFN-γ and the antiretroviral factors Apobec3G and SAMHD1. Gardiquimod (TLR7 agonist) did not appreciably alter levels of mRNA for IFN-γ, Apobec3G, or SAMHD1 at either 6 h or 24 h posttreatment when compared to untreated controls (Fig. 3, top panel). In contrast, the TLR8 agonist ssPolyU RNA induced significant decreases in IFN-γ expression at both the 6 h and 24 h time points as well as modest decreases in SAMHD1 expression at the 24 h time point (Fig. 3, middle panel). Activation of TLR9 using ODN2395 DNA resulted in a more than 6-fold increase in IFN-γ when measured 6 h posttreatment and a 3-fold increase after 24 h, and a 2- to 2.5-fold increase in Apobec3G and SAMHD1 at the 24 h time point (Fig. 3, lower panel). Data from each of the agonist-treated groups were normalized to expression levels of ribosomal protein L13A (RPL13A), and the level of each transcript was expressed relative to that measured in cells prior to the addition of agonist, and expression levels arbitrarily set to “1.”
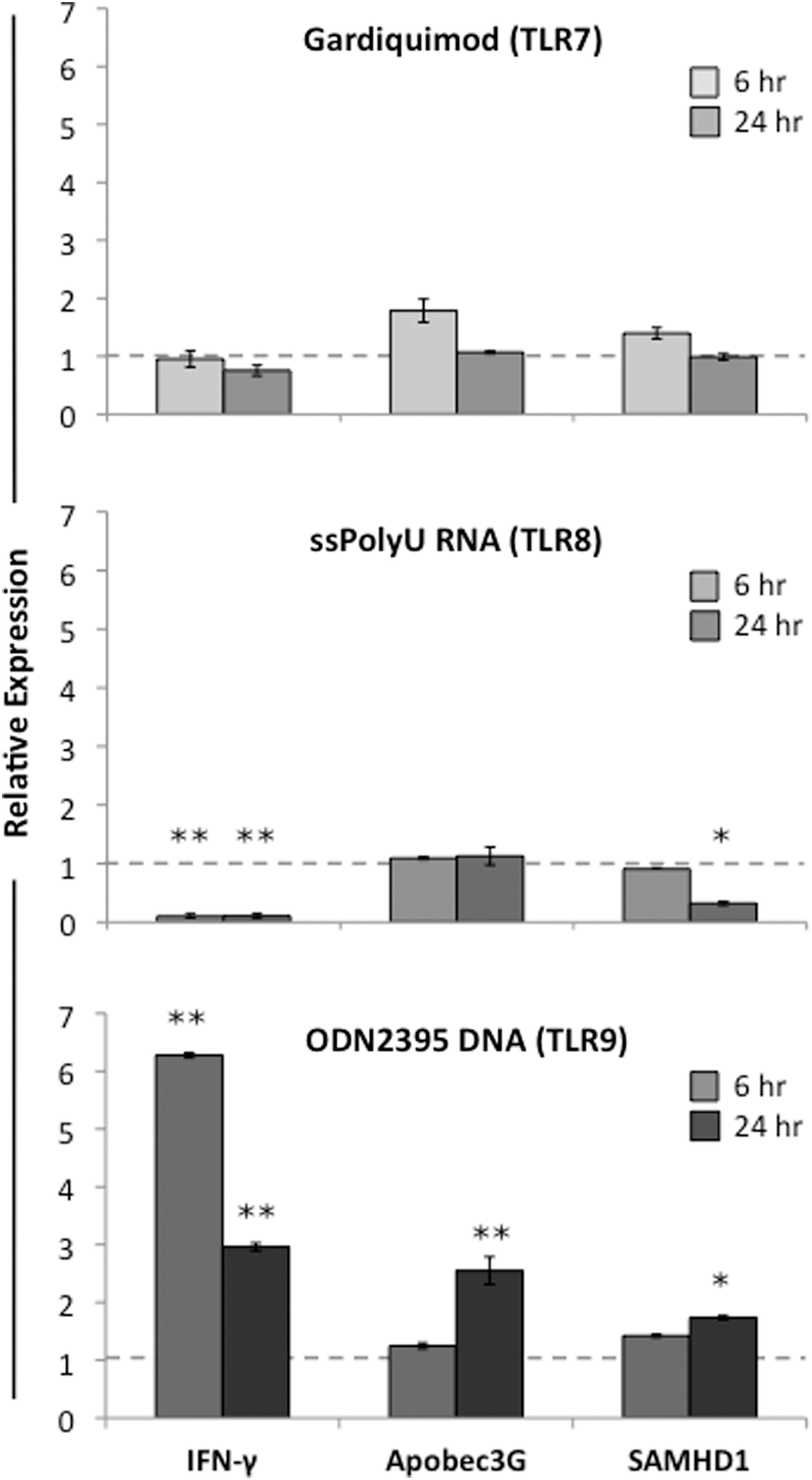
TLR9 activation induces type II interferon (IFN-γ) and retroviral restriction factors in PBMCs. PHA-activated PBMCs were cultured in triplicate wells in media alone or with 1 μg/ml gardiquimod (TLR7 agonist, upper panel), 0.156 μg/ml ssPolyU RNA (TLR8 agonist, middle panel), or 5 μg/ml ODN2395 DNA (TLR9 agonist, lower panel). After either 6 or 24 h incubation, cells were harvested and RNA was isolated for real-time PCR analysis of expression levels for IFN-γ and the retroviral restriction factors Apobec3G and SAMHD1. Transcript levels for these genes from the untreated controls at time 0 were arbitrarily set to “1” and are denoted by a dotted line. Tissue culture experiments were performed in triplicate using cells from three different donors; PCR experiments had six replicates per condition. *p<0.05; **p<0.005.
Inhibiting the myeloid differentiation primary response gene 88 (MyD88) blocks the induction of interferon following TLR8 activation, but enhances interferon gene expression in TLR9-activated cells
To determine the extent to which direct activation of TLR by agonist inhibited HIV-1 replication, a peptide that blocks the MyD88 adaptor protein was added to PHA-activated PBMCs prior to the addition of agonists to either TLR8 or TLR9. In the first analysis, measurement of transcript levels for type I (IFN-α and IFN-β) and type II interferons (IFN-γ) showed that while the MyD88 inhibitory peptide blocked the induction of type I interferons induced by agonist-mediated TLR8 activation, it enhanced both type I and type II interferon gene expression following TLR9 activation (Fig. 4A). In the second set of studies, PHA-activated PBMCs were first infected with HIV-1 and then incubated with the MyD88 inhibitor or the control peptide prior to agonist-mediated TLR activation using either ssPolyU RNA (TLR8) or ODN2395 (TLR9). In contrast to the findings with interferon gene expression, the MyD88 inhibitor peptide had no effect on the inhibition of HIV-1 replication following agonist addition (Fig. 4B).
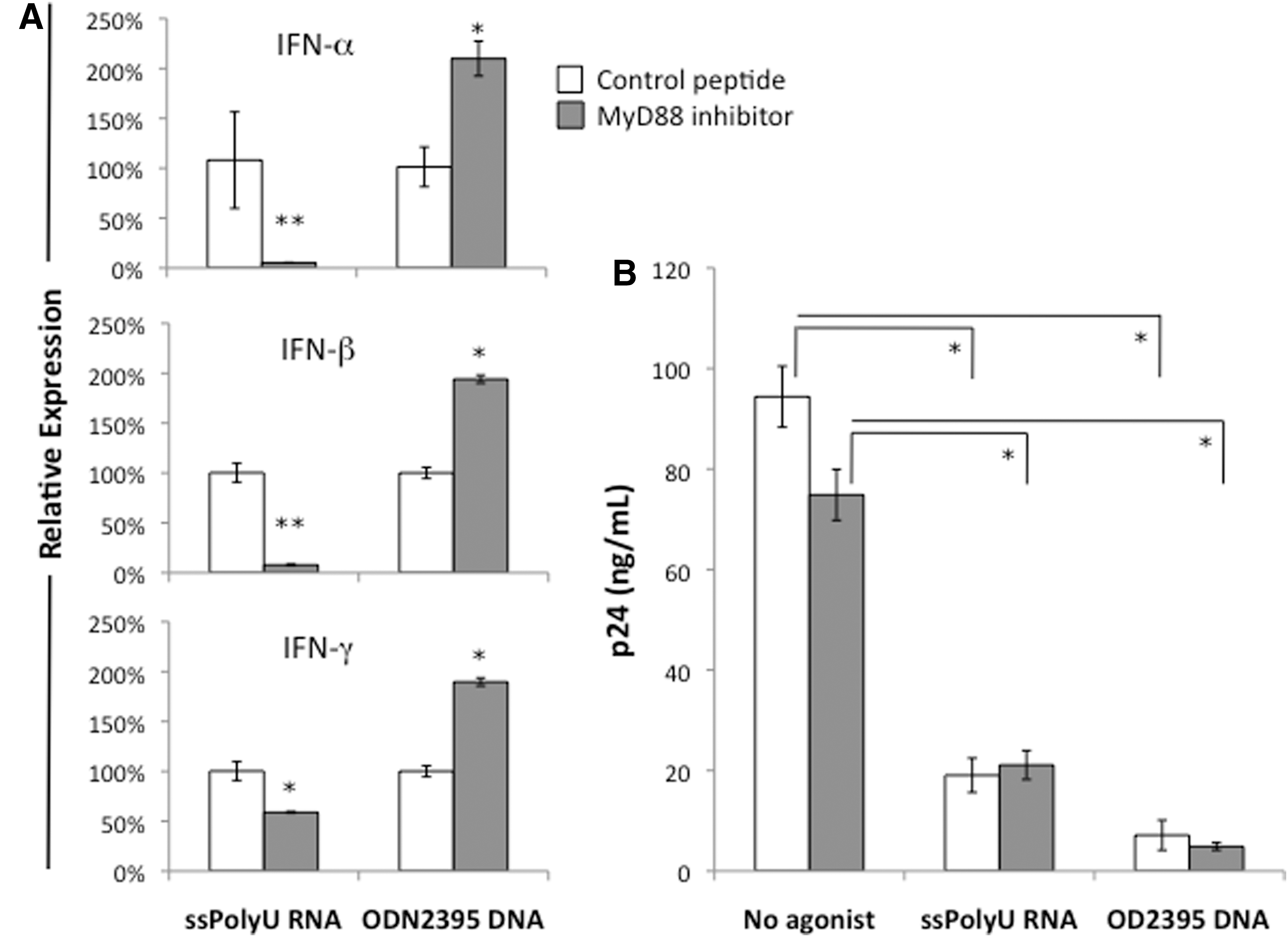
MyD88 inhibition blocks interferon expression in ssPolyU RNA-treated PBMCs but not ODN2395 DNA-treated cells, but does not reverse HIV-1 inhibition.
Sunitinib malate, a tyrosine kinase inhibitor, blocks TLR agonist-mediated inhibition of HIV-1
To assess the extent to which molecular events downstream of interferon gene expression contributed to the inhibition of HIV-1 replication, we used a small molecule kinase inhibitor, sunitinib malate, which has been reported to inhibit the interferon-induced antiviral proteins PKR and RNase L, 18 and assessed the effects of this inhibitor on HIV-1 replication. Pretreatment of HIV-1-infected PBMCs with sunitinib malate prior to TLR7, TLR8, or TLR9 activation with the appropriate agonist reversed the inhibition of HIV-1 replication (Fig. 5). In cells that did not receive a TLR agonist, sunitinib malate treatment alone did not alter HIV-1 p24 levels compared to cells that were treated with only DMSO control (Fig. 5).
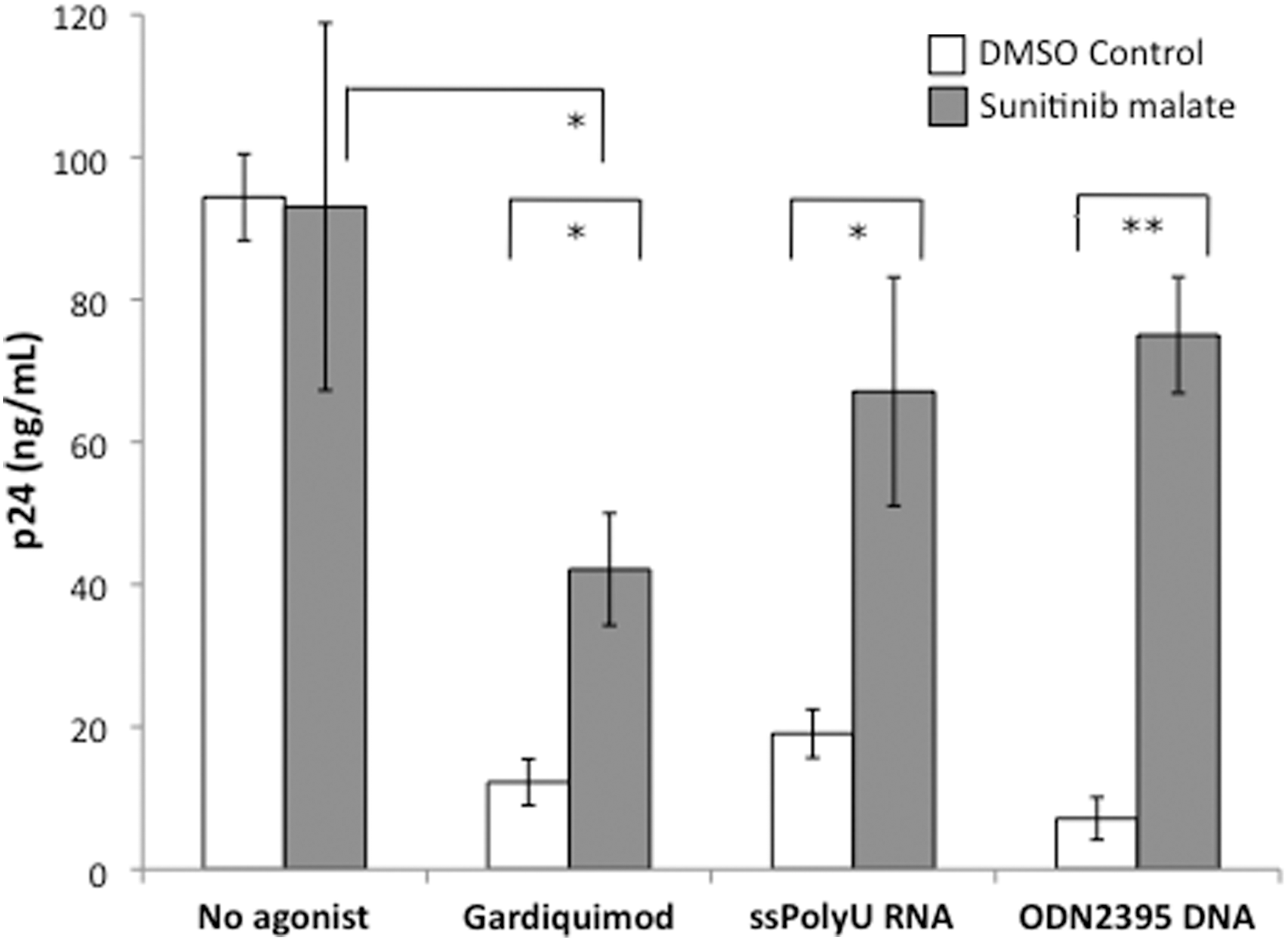
Sunitinib malate blocks TLR agonist-mediated inhibition of HIV-1. PHA-activated PBMCs were pretreated for 4 h with 5 μg/ml (10 μM) sunitinib malate. Inhibitor-treated PBMCs were infected with 20 TCID50 HIV-1BaL for 1 h, washed, then cultured for 6 days in media alone or in the presence of either 1 μg/ml gardiquimod, 0.156 μg/ml ssPolyU RNA, or 5 μg/ml ODN2395 CpG DNA before measurement of infection by HIV-1 p24 ELISA. All experiments were performed in triplicate using cells from three different donors. *p<0.05; **p<0.005.
Discussion
Toll-like receptors represent one of several classes of innate immune receptors that play an important role in protecting the host from pathogen invasion. These receptors, expressed primarily by leukocytes and mucosal epithelial cells, recognize unique molecules termed pathogen-related molecular patterns on bacteria, viruses, fungi, and protozoa. 31
The cellular expression of TLR varies among different leukocyte populations. For example, TLR3 is predominantly—if not exclusively—expressed by dendritic cells. 32 The finding that poly(I:C), a TLR3 agonist, was unable to induce a sustained anti-HIV effect in PBMCs was likely due to the absence of appreciable numbers of dendritic cells in the PBMC population. In contrast, TLR7, 8, and 9 are expressed on monocytes/macrophages, cells that constitute approximately 10–15% of the PBMC population. In addition, these TLR are also expressed on a subset of B cells that comprises about 20% of PBMCs. T cells have also been reported to express several of the TLR including TLR4, 5, 7, 8, and 9, where their expression regulates and limits immune responses to pathogens. 24,33 We observed a greater and more sustained anti-HIV effect in PBMCs activated with TLR8 and 9 agonists, and it is likely that the greater expression of these TLR on the majority of cells present in the PBMCs was in part responsible for this finding. It has also been reported that TLR9 ligands can stimulate replication of rat CD4+ T cells, 34 and so it is possible that the addition of the TLR agonist CpG DNA led to an increase in CD4+ T cell numbers throughout culture, enhancing the duration and magnitude of the antiviral effects. In fact, others have shown that TLR9 activation of T cells can protect these cells from apoptosis and prolong cell survival, 35 which could also increase the anti-HIV effects of these agonists in T cells.
Differences in anti-HIV activity among the agonists studied may also result from different patterns and/or concentrations of antiviral molecules induced by each agonist. Gardiquimod (TLR7), ssPolyU RNA (TLR8), and ODN2395 DNA (TLR9) induced high levels of the type I interferons, as well as the interferon-stimulated genes MxA, OAS, and PKR. Interestingly, only ssPolyU RNA induced significant levels of RNase L, a gene that codes for a potent nuclease that destroys both cellular and viral RNA molecules. 17 The degree to which each of these proteins contributed to the anti-HIV-1 effect is not known, but it is likely that the antiviral effect observed resulted from a combination of the types and quantities of known innate immune factors induced in PBMCs following TLR agonist addition.
Several groups, including ours, have shown that activating the TLR signaling pathways in immune cells using agonists and related small molecules potently inhibits HIV-1 infection and replication in activated lymphocytes and macrophages. 11,36 –39 Moreover, these same activation pathways have also been shown to increase immune responses to HIV-1 vaccines, primarily by inducing enhanced antibody responses. 40 –43 In contrast, other groups have shown that the addition of TLR agonists can, in some cases, lead to an increase in HIV-1 transmission and replication. 15 It is possible that in vitro studies would yield results differing from those performed in animal models or in patients due to the absence of immune cell recruitment or to interactions among differing cell types not represented in vitro.
We chose to define the cellular mechanisms by which PBMCs, composed primarily of lymphocytes and monocytes, control infection and replication of HIV-1 following TLR activation. By defining important host molecules that are potent HIV inhibitors, novel therapeutic agents could be designed to specifically induce these molecules in the absence of inflammatory responses. Such an effect could be achieved, for example, by the administration of a TLR8 or 9 agonist together with an MyD88 inhibitor. In this case, type I interferon production would be blocked, limiting the inflammatory response mediated by interferons, but at the same time inducing other anti-HIV molecules. In blood monocytes obtained from HIV-infected patients, TLR8 activation has been reported to suppress HIV-1 replication, perhaps indirectly via an increase in IL-12 production. 44 In another report, Campbell et al. showed that TLR8 ligands induced a vitamin D autophagic response in macrophages that led to an inhibition of HIV-1 replication. 39 Thus, the clinical utility of these agonists could be enhanced by the controlled and targeted application of these molecules in HIV-infected patients.
The addition of the tyrosine kinase inhibitor sunitinib malate reversed the anti-HIV activity of the agonists gardiquimod, ssPolyU RNA, and ODN2395. This tyrosine kinase inhibitor has been reported to inhibit the antiviral proteins RNase L and PKR 18 that are expressed farther downstream from the MyD88 adaptor protein. Because of the almost complete reversal of the HIV inhibition, this finding suggests that RNase L and PKR could be important molecules for HIV-1 inhibition. Sunitinib malate may inhibit the activity of PKR to a greater extent than that of RNase L, not only because the addition of the TLR9 agonist induced expression of PKR, but also because PKR may be a more effective anti-HIV molecule as it broadly inhibits viral replication even from integrated virus, whereas RNase L merely degrades viral RNA. Another possibility is that sunitinib malate blocks the signaling induced by the binding of cytokines to their receptors, thus blocking their antiviral effects. Of interest is that this inhibitor and others in its class are being used to treat non-AIDS-defining cancers that arise in HIV-infected patients. 45 Whether its use in HIV-infected patients with malignancies leads to an increase in viral replication is at present unknown, and suggests a close monitoring of plasma viral loads in these patients.
The addition of the MyD88 inhibitory peptide to PBMCs prior to treatment with ssPolyU RNA potently inhibited interferon induction, although there was no diminution in HIV-inhibitory activity. One likely explanation for these findings is that the ssPolyU RNA agonist served to stabilize the HIV-1 restriction factor SAMHD1, an enzyme that is responsible for restriction of HIV-1 replication in macrophages and myeloid dendritic cells. 46 In the absence of ssRNA, this enzyme complex is degraded. 22,46 An alternative explanation is that the HIV-1 activity observed in the absence of interferon production could have arisen from induction of retinoic acid inducible gene-1 (RIG-1), an alternative pattern recognition receptor that is a potent sensor of viruses. 47,48 Moreover, single-stranded RNA has also been reported to activate PKR by binding to the dsRNA recognition and triphosphate-binding site. 19
In contrast to the findings with ssPolyU RNA, the addition of the MyD88 inhibitory peptide prior to treatment with the agonist ODN2395 DNA actually increased the expression of both type I and type II interferons. Similar to the findings with ssPolyU RNA, the MyD88 inhibitory peptide was unable to block the anti-HIV-1 effect of ODN2395 DNA. These findings indicate that the induction of interferons by ssPolyU RNA is a direct result of TLR8 activation, whereas interferon production following ODN2395 DNA treatment is likely driven by mechanisms other than TLR9 activation. Although the induction of the type I interferons was observed for all agonists tested, only ODN2395 DNA (TLR9) treatment induced significant levels of the type II interferon, IFN-γ, and the enzymes Apobec3G, a cytidine deaminase that mutates cytidines to uracils during HIV-1 reverse transcription, 49 and SAMHD1, a phosphohydrolase that serves to deplete the nucleotide pool in the cytosol. 50 All of these molecules have potent anti-HIV activity, and probably were effective within hours of agonist addition to inhibit HIV-1 replication. It is also likely that the antiviral activity induced by ODN2395 DNA may not be mediated only via TLR9 activation, but may be activating alternative microbial DNA sensors in the cell. In support of this notion is the fact that the ODN2395 control, a segment of unmethylated DNA lacking both the palindromic sequence present in ODN2395 as well as the CpG motifs, was as effective as ODN2395 DNA in interferon induction and anti-HIV activity (data not shown). Although others have reported the anti-HIV activity of ODN2395 DNA, 36,37 these studies did not conclusively demonstrate that this agonist mediated anti-HIV effects directly through TLR9 rather than through other cytosolic DNA sensors. In fact, many alternative DNA sensors have been described that could account for the anti-HIV effect of ODN2395 DNA, including cyclic GMP-AMP synthase (cGAS), 51,52 the downstream protein stimulator of IFN genes (STING), 51,53,54 DNA-dependent activator of interferon regulatory factors (DAI), 55 and interferon-inducible protein 16 (IFI16). 56 These DNA sensors induce interferons in serine/threonine-protein kinase (TBK1)- and IRF3-dependent mechanisms similar to those employed by TLR3 signaling. 57,58 Moreover, these novel DNA sensors are broadly expressed by monocytes and lymphocytes, 59 in contrast to TLR9, which is expressed primarily in plasmacytoid dendritic cells and B cells, and thus they could function to potentiate a broader and more robust antiviral response in a larger proportion of the target cell population.
Our studies demonstrate that TLR agonists to endosomal TLR are highly effective in inhibiting HIV-1 replication through TLR-mediated as well as non-TLR-mediated mechanisms. The finding that blocking the signaling through TLR does not reverse the inhibition of HIV-1 replication indicates that alternative signaling pathways, most likely via cytoplasmic PRR, are contributing to the antiviral effects. It is therefore possible that these agonists could be developed as potent inhibitors of HIV-1 infection and replication, especially ODN2395 DNA, which inhibited HIV-1 replication even when added 72 h after HIV-1 infection. Moreover, the reported effects of TLR activation on enhancing adaptive vaccine responses to HIV-1 in mice 60 and nonhuman primates 61 indicate a dual purpose to these compounds in HIV-1 prevention. Thus, TLR agonists could be developed as novel therapeutic agents that serve both as adjuvants to boost vaccine responses as well as therapeutic reagents to inhibit viral transmission in HIV-1-exposed individuals. The inclusion of inhibitory peptides that block the induction of inflammatory cytokines such as interferons, yet retain anti-HIV effects, would be most beneficial in a clinical setting where inflammatory responses have been shown to enhance infection by HIV-1. In sum, our findings indicate that TLR agonists are powerful reagents that show promise as anti-HIV molecules by activating and promoting cellular pathways to block viral replication.
Footnotes
Author Disclosure Statement
No competing financial interests exist.