
Abstract
HIV-1 uses the coreceptors CCR5 and/or CXCR4 for cell entry. Monotropic CCR5-using variants are found early in the infection while CXCR4-using variants may appear after progression to AIDS. CXCR4 use may consist of both monotropic and dualtropic viruses. The viral phenotype is important in evaluating the response to CCR5 inhibitors, a new class of antiviral drugs. The coreceptor use of HIV-1 was investigated using population sequencing in 24 patients from Botswana, carrying HIV-1 subtype C and failing antiretroviral treatment, while 26 treatment-naive patients acted as controls. Single genome sequencing was used to discern minor HIV-1 populations in the treatment-experienced group. The Geno2Pheno method was employed to predict the coreceptor use phenotype from HIV-1 env gp120 V3 DNA sequences. The glycan-charge model adjusted for subtype C was also used for phenotype prediction. The viral phenotype of population sequences was predicted using Geno2Pheno in 24/24 treatment-experienced patients, of whom eight (33%) were predicted to harbor CXCR4-using strains as compared to 2/26 in the treatment-naive group (p=0.03). Single genome sequencing generated 4–23 clones/patient in the treatment-experienced group. Altogether, 90/295 (31%) putative CXCR4-using clones were identified. In 10/24 (42%) treated patients at least one clone was predicted to be CXCR4-using, further increasing the amount of identified treatment-experienced patients with CXCR4 use. Although subtype C is usually associated with comparatively little CXCR4 use, the frequency of CXCR4 use in treatment-experienced patients with subtype C can be higher, which may have implications for the administration of CCR5 inhibitors in this patient group.
Introduction
H
The prevalence of X4 strains in non-B HIV-1 subtypes varies. HIV-1 subtype D has been associated with an increased CXCR4 use, 12 –14 and some reports have suggested the same for subtype CRF01_AE. 15 –17 On the other hand, HIV-1 subtype C, which is the dominating subtype worldwide, 18 seems less likely to acquire CXCR4 use even at late clinical stages, 19 –22 perhaps because more mutations are required for a coreceptor switch. 23 However, later studies indicate that CXCR4 use in subtype C may not be as low as earlier reported, 24 –27 which could be a reflection of an evolving subtype C epidemic. 24,25,28
With the introduction of coreceptor inhibitors for the treatment of HIV-1, such as the CCR5 inhibitor maraviroc, which is currently in clinical use, 29 the analysis of the HIV-1 coreceptor use phenotype has become increasingly important for determining if this class of drugs can be used as part of the treatment regimen. Methods to determine if patients already harbor CXCR4-using strains, 29,30 which could have competitive advantages when CCR5 use is blocked, are therefore required. Conversely, no other class of drugs to date has been definitely correlated with preferred coreceptor use, 24,31 although some specific drug resistance mutations, such as the protease inhibitor-induced mutation L90M 24 or the nucleoside reverse transcriptase inhibitor-induced mutation M41L, 31 have been suggested to be related to increased CXCR4 use.
Subtype C-infected individuals who were failing antiretroviral treatment in Zimbabwe tended to harbor more CXCR4-using strains compared to untreated patients. 24 A similar observation was made in patients likely to carry subtype B in the United States 32 as well as in more recent studies from subtype C prevalent regions in South Africa. 26,27 During screening prior to a clinical trial of CCR5 inhibitor treatment among highly treatment-experienced patients, 50% of the subjects were found to harbor CXCR4-using virus populations. 30 Moreover, CXCR4-using viruses have been coupled with a higher prevalence of drug resistance mutations compared to R5 viruses in treatment-experienced patients. 33 Since maraviroc is mostly used as a salvage therapy drug, 34 it is critical to investigate further CXCR4 tropism in treatment-experienced patients, particularly those carrying subtype C for which CXCR4 use was reported to be less frequent.
The national HIV prevalence rate in Botswana is 17.6%, 35 with the majority carrying HIV-1 subtype C. Botswana offers free nationwide antiretroviral treatment for all HIV-infected patients who qualify for treatment under the national treatment guidelines since 2002. 36,37 Currently, up to 90% of all patients who qualify for treatment receive it 37 and the rate of treatment failure has been comparable to that in the developed world. 36
The overall aim of the current study was to investigate whether CCR5 inhibitors are suitable for treatment-experienced patients with subtype C. The specific aims were to compare subtype C coreceptor use in patients failing antiretroviral treatment in Botswana with treatment-naive individuals using population sequencing, as well as to further assess coreceptor use in treatment-experienced patients by analysis of single proviral genomes within the heterogeneous viral population of each treated individual.
Materials and Methods
Patients and samples
The samples were collected from all adult patients experiencing second-line treatment failure in Botswana's national antiretroviral treatment (ART) program over a 2-month period (May to June) in 2006. Twenty-four patients were identified in total, and both sexes were represented in the material. The patients were on treatment for a minimum of 1 year (the exact duration was unavailable) and virological failure was determined as two consecutive viral load measures above 1,000 RNA copies/ml, which enabled drug resistance genotyping.
The actual treatment regimen provided was not known, but at the time of sample collection the standard first-line treatment consisted of two nucleoside reverse transcriptase inhibitors (NRTIs), zidovudine (AZT) and lamivudine (3TC), together with one nonnucleoside reverse transcriptase inhibitor (NNRTI), either nevirapine (NVP) or efavirenz (EFV), while second-line treatment usually consisted of two other NRTIs, didanosine (ddI) and stavudine (d4t), and one protease inhibitor (PI), nelfinavir (NFV). 36 The majority if not all of the 24 patients were failing the second-line regimen, although there could be one or two who were coming from the private sector who might have been exposed to different drugs.
Four milliliters of peripheral blood was collected in ethylenediaminetetraacetic acid (EDTA)-coated tubes from each patient. The blood was centrifuged to separate plasma from cells. The plasma was either directly used for drug resistance testing or stored at −70°C, alongside the buffy coat. The drug resistance testing covered all known PI, NRTI, and NNRTI mutations, and was performed as previously described.
38
Drug resistance profiles were available for 12 of the patients (Supplementary Table S1; Supplementary Data are available online at
Twenty-six treatment-naive patients infected with subtype C were selected as controls from a previously described cohort with samples collected in 2003. 39 As the national antiretroviral treatment program in Botswana was initiated in 2002, 36,37 it would be likely that the treatment failure patients were started on treatment at a time point similar to the collection date of the treatment-naive control samples. Also, the likelihood of finding treatment-naive patients with low CD4 counts would be higher in a cohort from an earlier time point, since the guidelines for commencement of treatment were different at the time. The inclusion criteria for the controls were an available envelope sequence spanning the V3 region of glycoprotein 120 and a CD4 count of 250 cells/μl or less, which would have made these patients eligible for treatment in 2006.
DNA extraction and polymerase chain reaction (PCR)
DNA was extracted from 200 μl of buffy coat, using the QIAamp Blood kit (Qiagen, Chatsworth, CA), eluted in 200 μl and stored at −20°C until ready for use. Sequences corresponding to the HIV-1 envelope (env) glycoprotein (gp) 120 V3 regions and flanking C2-C3 regions were amplified by nested PCR. The primers for the first round PCR were JA167 and JA170, and for the second round were JA168 and JA169. 40 Each PCR round consisted of 40 cycles and two PCR rounds were performed. 41
Limiting dilution and single V3 genome sequencing PCR
For the 24 patients failing treatment, limiting dilution PCR followed by single genome PCR were performed to generate molecular clones. In addition, population sequencing of the DNA samples was carried out. The DNA samples from the control patients were obtained from a previous study, where only population sequencing had been performed. 39
Limiting dilution PCR was run in 4-fold dilutions of the sample DNA with four replicas of each dilution, starting at 1:8, in MicroAmp Optical 96-well reaction plates (AB Applied Biosystems, Singapore). A dilution was calculated, which would yield ≈25% positive reactions. A total of 40–48 PCR replicas were run using the determined dilution. This would yield a high proportion of PCR products from single genomes of the various viral cDNA copies present in the patient peripheral blood.
DNA sequencing, editing of chromatograms, and generating phylogenetic trees
PCR products were purified using the QIA quick PCR purification kit (QIAGEN GmbH, Hilden, Germany) when running PCR in tubes, and the Multiscreen PCR 96 Montage Life Science kit (Millipore Corporation, Bedford, MA) when running PCR in 96-well plates (AB Applied Biosystems, Singapore). Of the purified PCR product 10 ng/μl (Nanodrop) was used in a 20 μl Big Dye terminator 3.1 (Applied Biosystems, Foster City, CA) sequencing reaction. The primers JA168 and JA169 (3.0 pmol/μl each) were used for population sequences, while molecular clone sequences were often generated with one primer only. Plates were sent to Eurofins MWG Operon, Ebersberg, Germany for sequencing.
The sequences were edited and assembled into overlapping fragments using the software program Sequencher (Gene Codes Corporation, Ann Arbor, MI). Each sequence was analyzed for ambiguities at the nucleic acid level of the full product and amino acid level of the V3 region, respectively. The majority rule was used for the V3 region amino acid sequence determination of population sequences. Ambiguity codes were used for determination of sequences analyzed in phylogenetic trees.
Consensus sequences were aligned with Clustal X version 1.81 (
A sequence would be regarded as a molecular clone if ambiguities were completely absent from V3. If no more than one ambiguity within V3 was present, the sequence would represent two molecular clones and both would be included for the phylogenetic analysis. Ambiguities in the flanking regions were nevertheless allowed, meaning that each molecular clone would be V3 unique, but could consist of multiple variants sharing the same V3 nucleotide sequence.
Sequences spanning the same region from the control patients
39
(see Sequence Data for accession numbers) were extracted from the Los Alamos HIV Sequence Database (
Phenotype characterization
The phenotype was predicted using two bioinformatic methods, the Geno2Pheno [coreceptor] method
42
(
The phenotype was also predicted using a glycan-charge model. 43,44 Sequences containing the combination of the V3 region glycosylation motif at position N301 and total V3 charge below 5.0 were regarded as R5, while sequences with a total V3 charge above 5.1 were regarded as X4. Sequences containing the glycan motif and a total charge of 5.0–5.1 were regarded as mixed, i.e., R5, X4, or dualtropic could not be discerned from one another in this group. The charge cut-offs were based on subtype C. 44
Statistical methods
Two-sided Fisher's exact test was applied when comparing the outcome between the treatment-naive and treatment failure group, using the same method (population sequencing). The two-sided exact sign test was applied when comparing different methods (population versus single genome sequencing) within the treatment failure group only. Two-sided Student's t-test was applied when comparing the genetic distances from the root of the phylogenetic tree between R5 strains and CXCR4-using strains.
Ethical permission
The study was approved by the ethical committee in Botswana. The regional board of ethical vetting in Stockholm also approved of it with the registration number 2008:2007/1496-31/3. All the enrolled patients provided informed consent.
Results
Generation of HIV-1 sequences from the treatment failure patients
We generated 24 population sequences from 24 patients (Fig. 1A) and 295 molecular clone sequences. There were 4–23 molecular clones per patient (Table 1).

Alignment of the V3 region population sequences from patients failing antiretroviral treatment
DT, dualtropic.
Population sequences of treatment-naive patients versus treated patients
The samples for the treatment-naive group have been described previously, 39 and their sequences are presented in Fig. 1B. Here, individuals who would qualify for treatment by the standards of the government of Botswana of 2006 were selected. Twenty-six patients with CD4 counts below or equal to 250 cells/μl qualified to be included in the analysis. Of the 26 sequences, two were predicted to be from viruses able to use CXCR4 by the Geno2Pheno method, while the glycan-charge model predicted all 26 to be R5 (Table 2).
The mixed group is regarded as CXCR4-using.
Two-tailed Fisher's exact test.
Two-tailed exact sign test.
European recom. cut-off=recommendations from the European Consensus Group on clinical management of HIV-1 tropism testing (10% false-positive rate).
All 24 samples were amplified directly from the DNA of patients failing treatment, although the population sequences for a few were obtained after performing several replicas of PCR or after dilution of the original DNA sample. An alignment of the V3 loop amino acid population sequences is shown in Fig. 1. The Geno2Pheno phenotype prediction found eight of the sequences to be from CXCR4-using viruses while the remaining sequences were from CCR5-using viruses. The glycan-charge model predicted five sequences to be potential CXCR4 users and the remaining to be CCR5 users (Table 2).
The seemingly higher proportion of CXCR4-using viruses in the treated individuals' populations (8/24, 33.3%) versus the treatment-naive individuals'populations (2/26, 7.7%) as predicted by Geno2Pheno was statistically significant (p=0.03; Table 2). This was also the case for the predictions made by the glycan-charge model: 5/24 (20.8%) treated CXCR4 users versus 0/26 (0%) treatment-naive users (p=0.02; Table 2).
Single genome sequencing of the V3 region and phenotype prediction
Single genome sequencing was used to identify potential CXCR4-using viruses that were not detected by population sequencing in the treatment failure patients. The information in Table 1 shows the phenotype predictions of the molecular clones by the Geno2Pheno method and the glycan-charge model in these patients. Using Geno2Pheno, 10 (41.7%) of the 24 patients had at least one molecular clone that was predicted to use CXCR4, while the other 14 had clones predicted to use CCR5. Altogether 90/295 (31%) of the clones were characterized as putatively CXCR4-using.
In two patients with sample numbers 2 and 13, the proportion of CXCR4-using molecular clones was low, with a clear risk of misdiagnosis if based solely upon population sequences (Fig. 1A). In each case at least one clone was detected to be able to use CXCR4 (Table 1). CXCR4-using strains could be detected at a level of ≥7% (1 of 14) using single genome sequencing (Table 1). All eight patients predicted to harbor X4 or R5X4 strains by population sequencing had CXCR4-using clones (sample numbers 1, 5, 6, 9, 17, 21, 22, and 23).
Using the glycan-charge model, 16 individuals (66.7%) of the 24 patients had at least one molecular clone predicted to be potentially CXCR4-using, while the remaining eight were considered to be R5. The mixed group can contain R5, X4, and dualtropic strains, but for the purpose of not risking missing potential CXCR4 users, patients who had sequences in this category were all considered as possible carriers of CXCR4-using virus. Hence, altogether 68/295 (23%) of the molecular clones were characterized as potential CXCR4 users (out of which 29 were in the mixed category).
By population sequencing (Fig. 1A), which detects CXCR4-using virus only if the frequency is ≥25%, there would be a clear risk of misdiagnosis of 11 patients. For seven cases, at least one clone was classified as a CXCR4 user, while for the remaining four cases at least one clone was in the mixed group (Table 1). CXCR4-using strains could be detected at a level of ≥8% (1 of 13) using single genome sequencing, decreasing this further to ≥7% (1 of 14) if molecular clones from the mixed group were regarded as CXCR4-using (Table 1). All five patients predicted to have X4 or classified into the mixed group by population sequencing harbored X4 or mixed clones as expected (sample numbers 1, 5, 17, 20, and 22).
The prevalence of CXCR4-using HIV-1 strains determined by population sequencing was compared to prevalence based on single genome sequencing (Table 2). Single genome sequencing revealed more CXCR4-using strains than population sequencing, which was statistically significant for the glycan-charge model predictions (p=0.001), but not for the Geno2Pheno predictions (p=0.5).
Phylogenetic relationship of the molecular clones
All molecular clones and population sequences were analyzed for their phylogenetic relationships as shown in Fig. 2A. The viral molecular clones from each patient were well separated into individual clusters (bootstrap values of 71 or above), except the sequences of patient 8 and 12 for whom bootstrap values were lower. Furthermore, the sequences of patient 2 formed two distinctly separate (from each other and from all other patients) clusters, possibly due to a superinfection.
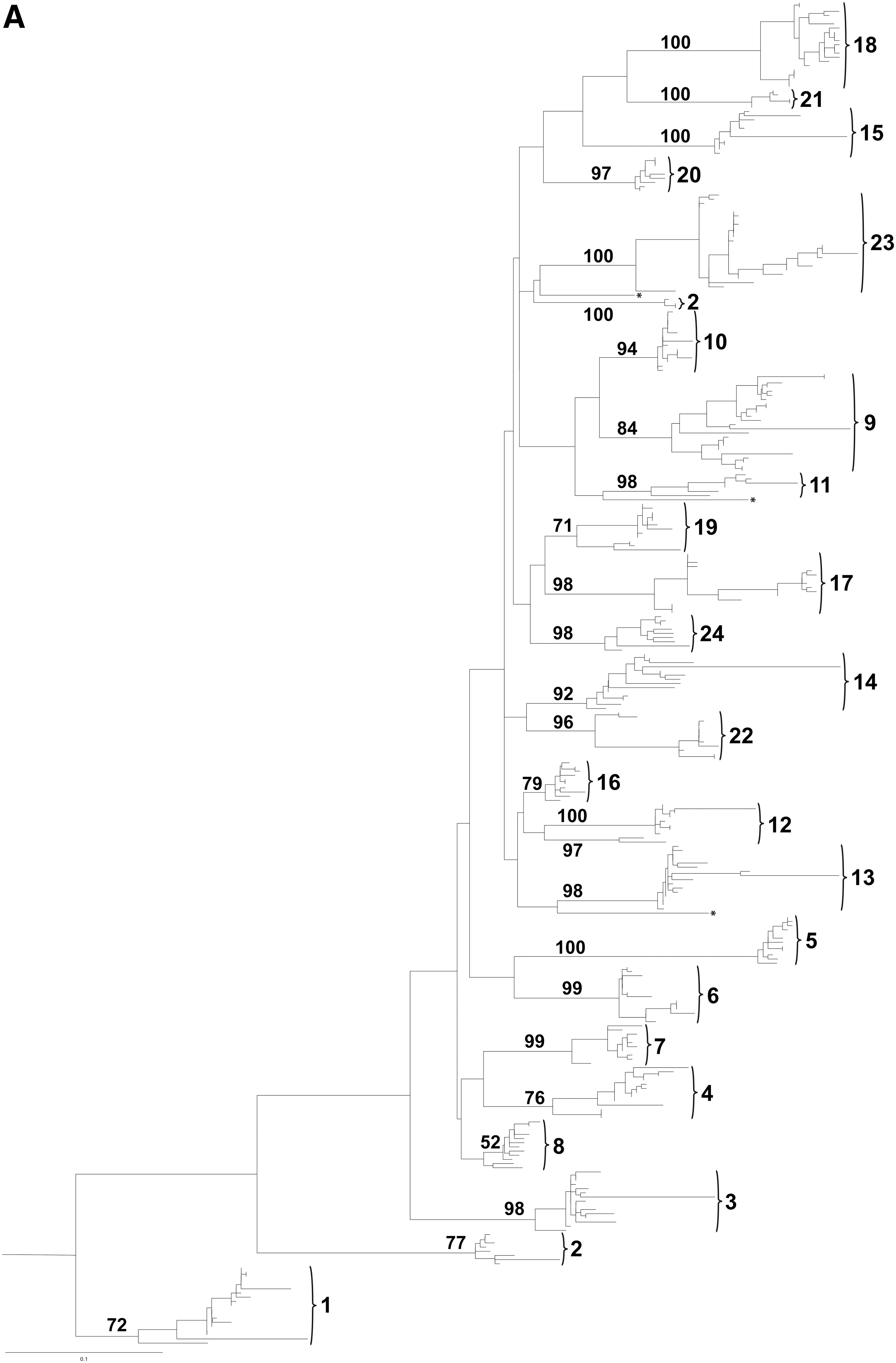
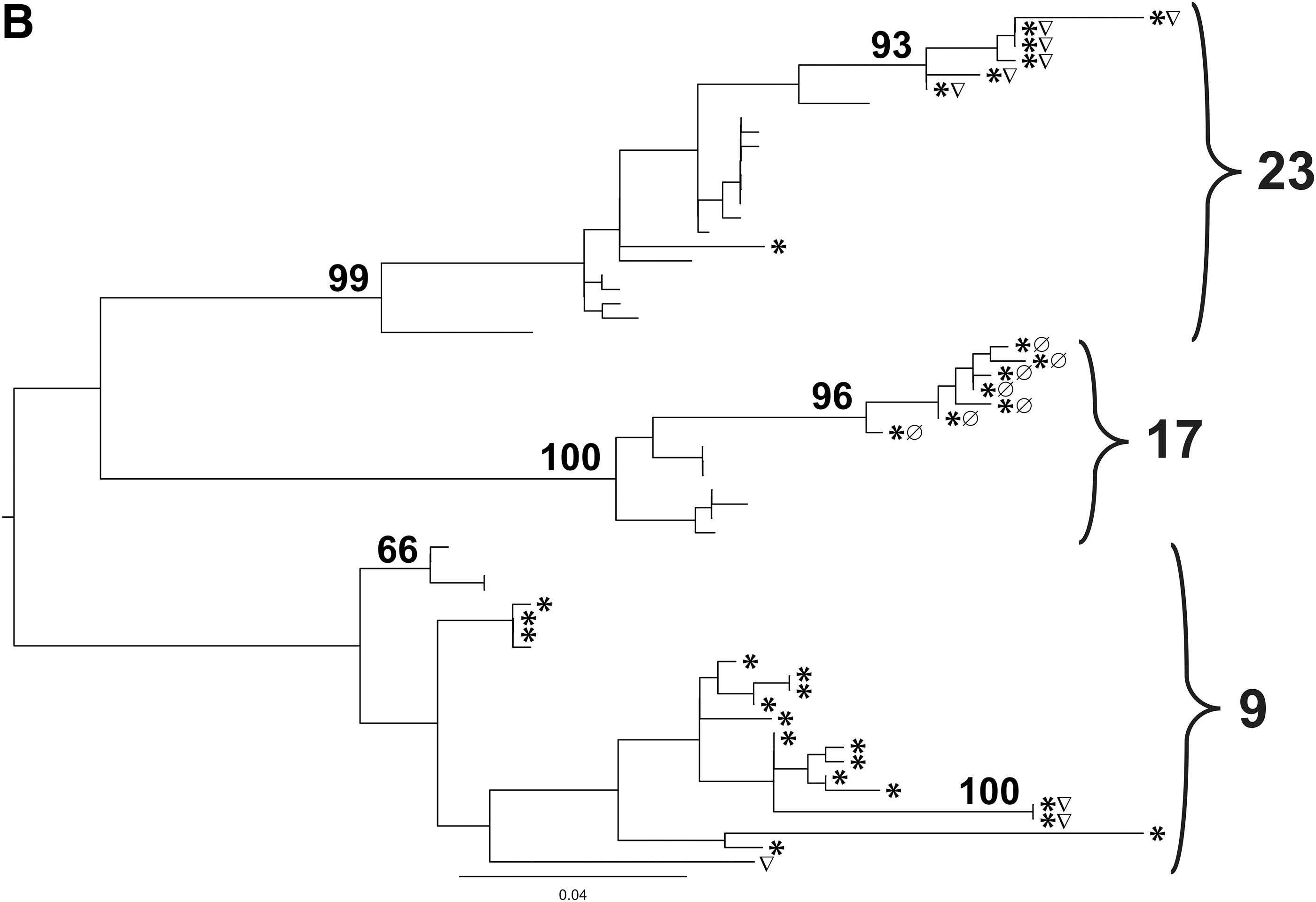
Figure 2B shows the phylogenetic tree of patients 9, 17, and 23, who harbored several CCR5- and CXCR4-using molecular clones. For all three individuals, there was a tendency for the molecular clones to cluster by coreceptor use. There was also a clear tendency that CXCR4-using strains had longer genetic distances from the root of the tree compared to the R5 strains, which was statistically significant for all three patients: 9, 17, and 23. The probability (p) in each case was 1.88×10–3, 5.8×10–4, and 1.5×10–6, respectively, for Geno2Pheno-based predictions, and 4.34×10–2, 1.0×10–8, and 4.0×10–8, respectively, for glycan-charge-based predictions (Student's t-test), rendering both Geno2Pheno and the glycan-charge model's coreceptor use predictions plausible, even when they did not overlap.
Discussion
In patients infected with HIV-1 subtype C and failing antiretroviral treatment, we detected a statistically significant increased frequency of CXCR4-using viruses by genotypic tropism testing on population sequences than in treatment-naive patients, suggesting that CCR5 inhibitors may be less suitable as drugs in treatment-experienced as opposed to in treatment-naive patients. An even higher frequency of CXCR4-using viruses was detected using single genome sequencing in treatment-experienced patients.
Suboptimal treatment 24 and specific drug resistance mutations such as L90M 24 and M41L 31 have been suggested to create a favorable environment for the CXCR4-using phenotype. However, in Botswana the treatment adherence rates have been good, 36 and no statistically significant correlation could be observed in this study between coreceptor tropism and specific drug resistance mutations or drug classes, although it should be noted that only a smaller subset of the patients was available for this analysis (Supplementary Table S1). Low CD4 counts were previously suggested as an explanation for the increased presence of CXCR4-using viruses in treatment-experienced patients. 24,26,27 In the current study, the treatment-naive controls had CD4 counts that would have warranted treatment, and hence likely matched the CD4 counts of the treatment-experienced patients, although it cannot be excluded that the treatment-experienced patients had even lower CD4 counts at treatment initiation. However, the collection date was later for the treatment-experienced patients compared to the controls (2006 and 2003, respectively), suggesting that the higher CXCR4 use could be the result of the evolving subtype C epidemic. 24,25,28
Interestingly, a large recent study in Botswana focusing on treatment-naive women infected with subtype C with low CD4 counts showed that the prevalence of CXCR4-using virus was still low in this group 46 compared to the prevalence in subtype B-infected individuals. In a subset of that study comparing coreceptor use phenotype before and after treatment, CXCR4-using viruses emerged in patients after treatment failure, albeit at lower frequencies than in the current study. This could perhaps be attributed to the fact that the patients studied here were on second-line treatment, suggesting that they may have had more opportunity to acquire multiple drug resistance mutations, which has been linked to CXCR4 use. 33 Thus, a limitation of the study was that patients failing first-line antiretroviral treatment could not be included, since according to the national treatment guidelines, only patients failing second-line treatment are entitled to drug resistance genotyping. As the source of samples was the national drug resistance genotyping laboratory, only samples where genotyping had been performed for clinical reasons could consequently be accessed.
Maraviroc, the only CCR5 inhibitor in clinical use to date, is approved by the U.S. Food and Drug Administration (FDA) for inclusion in treatment regimens both for treatment-experienced 29,34 and treatment-naive patients, 34 but it is rarely used as a first-line drug due to the twice-daily dosing schedule and the requirement of coreceptor use phenotype determination prior to administration. 34 In the United States, the costly enhanced sensitivity Trofile assay for coreceptor use determination needs to be performed before starting a patient on maraviroc. In contrast, in Europe genotypic assays, where coreceptor use phenotypes are predicted based on V3 population sequences, are recommended, in particular the Geno2Pheno algorithm with a 10% false-positive rate, 45 used in the current study. However, phenotype prediction algorithms with equivalent or more accurate predictions that are subtype-specific are still sought, such as the adjusted glycan-charge model presented here.
The source of material for genotypic assays can be either viral RNA from plasma or proviral DNA from peripheral blood mononuclear cells (PBMCs). Virus carrying drug-resistant mutations has an increased replicative capacity in the presence of drugs and is detectable in plasma before it can be found in PBMCs. 47 The tropism testing in this study was performed on proviral DNA, which could affect the relevance of the tropism results to the circulating drug-resistant virus. However, studies have shown high concordance between tropism predictions made from RNA and DNA compared to phenotypic assays in the same treatment-naive and treatment-experienced patients. 48,49 Moreover, genotypic tropism testing on proviral DNA compared to viral RNA possibly increases the chance of identifying CXCR4-using virus, 49,50 and can be performed on patients with suppressed or undetectable viral load, 51 signifying its potential clinical usefulness.
There are various methods to obtain sequences for genotypic assays. Population sequencing is an easily accessible, fast, and fairly inexpensive method. However, the sensitivity of population sequencing is limited as only frequencies above 25% of the total viral strain population are detected. Single genome/clonal sequencing methods can detect strains that make up less than 25% of the total viral quasispecies, and have for instance improved the sensitivity in detecting nevirapine resistance mutations. 52 Single genomes can be obtained using bacterial cloning techniques or, as in this study, through limiting dilution PCR. Bacterial cloning methods can give more reliable results in terms of confirming that the sequences belong to single viral strains, but have a higher cost and are more labor intensive, whereas single genome sequencing based on limiting dilution PCR provides a similar and, in some aspects, better measure of population diversity compared to bacterial cloning techniques. 53 In the present study, using limiting dilution PCR, the range of putative CXCR4-using strains detected in a single patient varied between 7% and 100%. To detect CXCR4-using strains at lower levels, either more single genome sequences would need to be generated or other methods such as the more elaborate and expensive pyrosequencing, 54,55 where a very large number of clones can be detected simultaneously from the same sample, would need to be used, although also here the costs and labor intensity might restrict the number of patients that it would be possible to include, especially in resource-limited settings.
A limitation of this study was the unavailability of material for creating single genomes from the treatment-naive patients, since only population sequence data from 2003 were available for the treatment-naive patients and not their DNA samples. Hence, a comparison between the treatment-naive and the treatment-experienced patients could be performed only on a population sequencing level, ruling out the ability to assess how the potential existence of minor CXCR4-using populations in the treatment-naive group would affect the comparison with the treatment-experienced group, where we know more CXCR4 users were detected using single genome sequencing. Nevertheless, the clinical impact of small proportions of CXCR4-using virus among a patient's quasispecies is not known. It is possible that the consequence of harboring CXCR4-using virus is affected by disease stage, since CXCR4-using virus is more easily neutralized by an immunocompetent individual. 28 Prospective studies are needed to learn more about the disease outcome in such patients, since the outcome may influence the choice of method for identifying CXCR4-using strains.
Here, the treatment-experienced patients did not receive CCR5 inhibitors. Still, many CXCR4-using sequences were present, which may challenge the use of CCR5 inhibitors in treatment-experienced patients with subtype C. The low frequency of CXCR4-using variants recently reported in treatment-naive patients with advanced disease, 46 and shown here, further supports the usefulness of maraviroc as a first-line drug among subtype C patients, at least in Botswana. If maraviroc is to be used for patients failing treatment in this setting, it would be strongly recommended that viral phenotype prediction be conducted first to ensure susceptibility of the virus to the drug. Further studies are warranted that address CXCR4 use in treatment-naive and treatment-experienced patients on a higher scale using both population sequencing and single genome analysis, as well as clinical trials that compare treatment outcomes and drug resistance development in patients treated with CCR5 inhibitors within salvage therapy regimens compared to within first-line treatment regimens.
Sequence Data
The accession numbers for the DNA sequences of the 26 patients used as controls were AY887852-5, AY887857-64, AY887866-72, AY887874-6, AY887879-80, AY887882, and AY887884. The accession numbers for the DNA sequences of the 24 drug-resistant patients were JN808454–JN808772. The accession numbers for the subtype C reference sequences were AY772699, U46016, and U52953.
Footnotes
Acknowledgments
We gratefully acknowledge all the patients included in the study. We would also like to thank Professor Gunilla Karlsson Hedestam for reviewing the manuscript. This work was supported by an MD-PhD training grant from the Karolinska Institutet to L.P.S., an EU Marie Curie Early Training grant for S.G., and a grant from SIDA to A.E.
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.