
Abstract
Elite controllers or suppressors (ES) control viral replication without antiretroviral therapy. While many ES are infected with replication-competent virus, others have evidence of infection with attenuated isolates. Here we report a case of an ES infected with an HIV-1 isolate that contained a 38-base pair deletion in nef that led to a reading frame shift and a premature stop codon. Interestingly, clones amplified from plasma or cultured from CD4+ T cells between 2006 and 2008 contained one of two separate compensatory deletions that restored the reading frame. A new insertion generated by duplication of adjacent sequences was found in isolates obtained in 2010 and this evolution was accompanied by the development of low level viremia. This article provides evidence of the evolution of an attenuated HIV-1 isolate toward greater virulence in an elite suppressor.
H
The mechanisms by which patients control viral replication are poorly understood, and the roles that host and viral factors have in determining the clinical outcome of HIV-1 infection are debated. Initially, infection with a defective virus was hypothesized to result in long-term control of viral replication. Strong evidence for this came from the Sydney Blood Bank Cohort, in which seven patients were infected by transfusion transmission of HIV-1 from blood of a single donor. All patients were observed to have a common deletion in nef and the U3 LTR. 2 Nef is important for HIV-1 replication and is essential for SIV infection, 3 and subsequent studies demonstrated that deletion in nef and other accessory genes was associated with long-term control of HIV-1. 4,5 Various studies that relied on the analysis of HIV-1 proviral sequences indicated that virus amplified from some ES and VC had large insertions and deletions or difficult to revert polymorphisms in essential genes. 6,7
However, subsequent studies indicated that some ES were infected with replication-competent virus that replicated well in vitro. 8 No large deletions or insertions were observed in full HIV-1 genome sequence analysis 8 ; furthermore, transmission pairs studies indicated that ES were able to control virus that caused progressive disease in other individuals. 9 Multiple studies have indicated that HLA-B*57 is overrepresented in ES cohorts and that domains in the HLA locus are most associated with control in genome-wide association studies (GWAS). 1 Additionally, a qualitatively superior CD8+ T cell response has been implicated in elite control, 10 further suggesting a dominant role of host immune factors in determining clinical outcome of infection. Thus, while infection with defective virus may play some role in the control of viral infection, it is clear that some ES are able to control full pathogenic virus.
Herein, we describe an ES who maintained elite control for greater than 10 years who was most likely infected with an attenuated virus. Sequence analysis revealed a defective nef gene, but viral evolution occurred and was associated with the development of low level viremia. These data suggest that some viruses with large deletions in nef are able to continue to replicate and evolve.
ES11 is an African American female with a history of injection drug use who tested positive for HIV-1 infection at the age of 50. Her first recorded CD4 count was 782 cells/μl and her first viral load was <400 HIV-1 RNA copies/ml of plasma. She maintained undetectable viral loads for 10 years but developed persistent low-level viremia 5 years ago (Fig. 1). To determine the cause of elite control in this patient virologic analysis was performed.
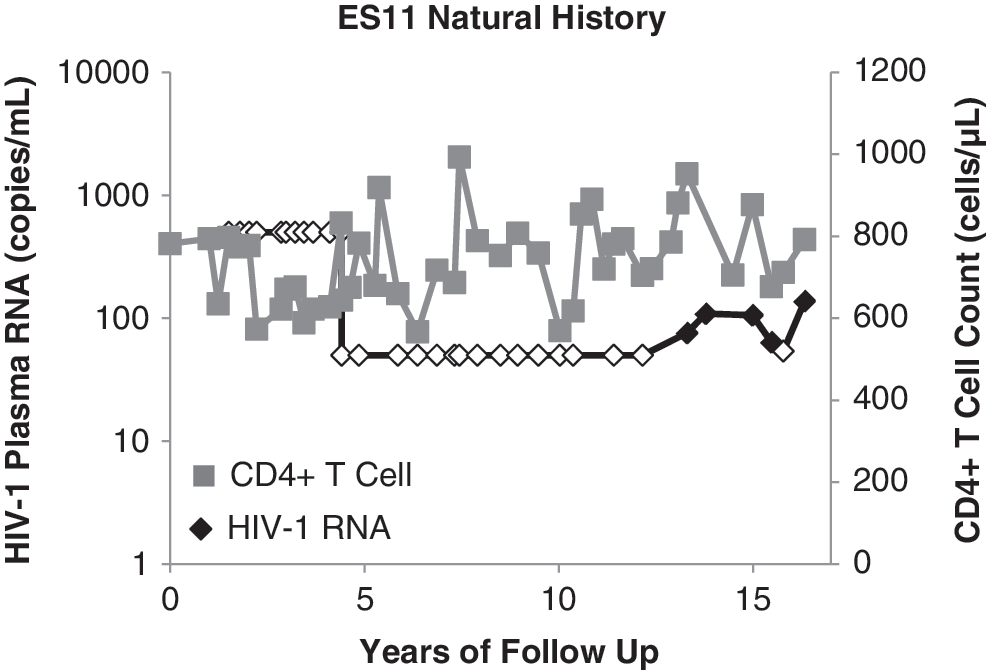
Patient natural history of infection. The natural history of infection for ES11 is shown over years of follow-up. CD4+ T cell counts (gray squares) in cells per μl and HIV-1 plasma RNA (black diamonds) in copies per ml are indicated on the y-axis. Undetectable HIV-1 plasma RNA levels are indicated by an open diamond.
Informed consent was obtained prior to the study. Handling of all patient samples was done in accordance with the regulations of the Johns Hopkins University Department of Medicine.
Previous studies have suggested that viral fitness could be a determining factor in the control of viral replication. Thus, to determine the fitness of the infecting virus, we used a sensitive coculture assay to culture primary viral isolates from the latent reservoir to use in a fitness assay. 8
To analyze viral fitness, peripheral blood mononuclear cells (PBMCs) from healthy donors were cultured for 2 days in the presence of interleukin (IL)-2 and phytohemagglutinin (PHA). CD4+ T cells were isolated using the CD4+ T cell isolation Kit II (Miltenyi) and infected by spinoculation with 200 ng of p24 of two representative patient isolates, the laboratory isolates Ba-L and IIIB, or other previously described primary isolates from two chronic progressors (CP1 and CP29) and one viremic controller and one ES (ES38 and VC19) (Fig. 2A). Supernatant samples were taken over the course of 10 days. By day 10 postinfection, there was more than a log difference in the amount of p24 produced by CD4+ T cells infected with isolates from ES11 compared to cells infected with both reference strains and representative primary isolates from CP and controllers (Fig. 2A). At the end of the first round of infection, culture supernant was collected and used immediately to infect PHA-activated CD4+ T cells from a second healthy donor (Fig. 2B). Equal quantities of virus, as determined by p24 concentration, were used for the second round of infection and infection was performed at 20 ng of p24 per million CD4+ T cells. Supernatant samples were again taken over the course of 10 days. Viral replication was quantified by p24 ELISA (Perkin Elmer). The same deficiency in replication was observed in the second round fitness assay.
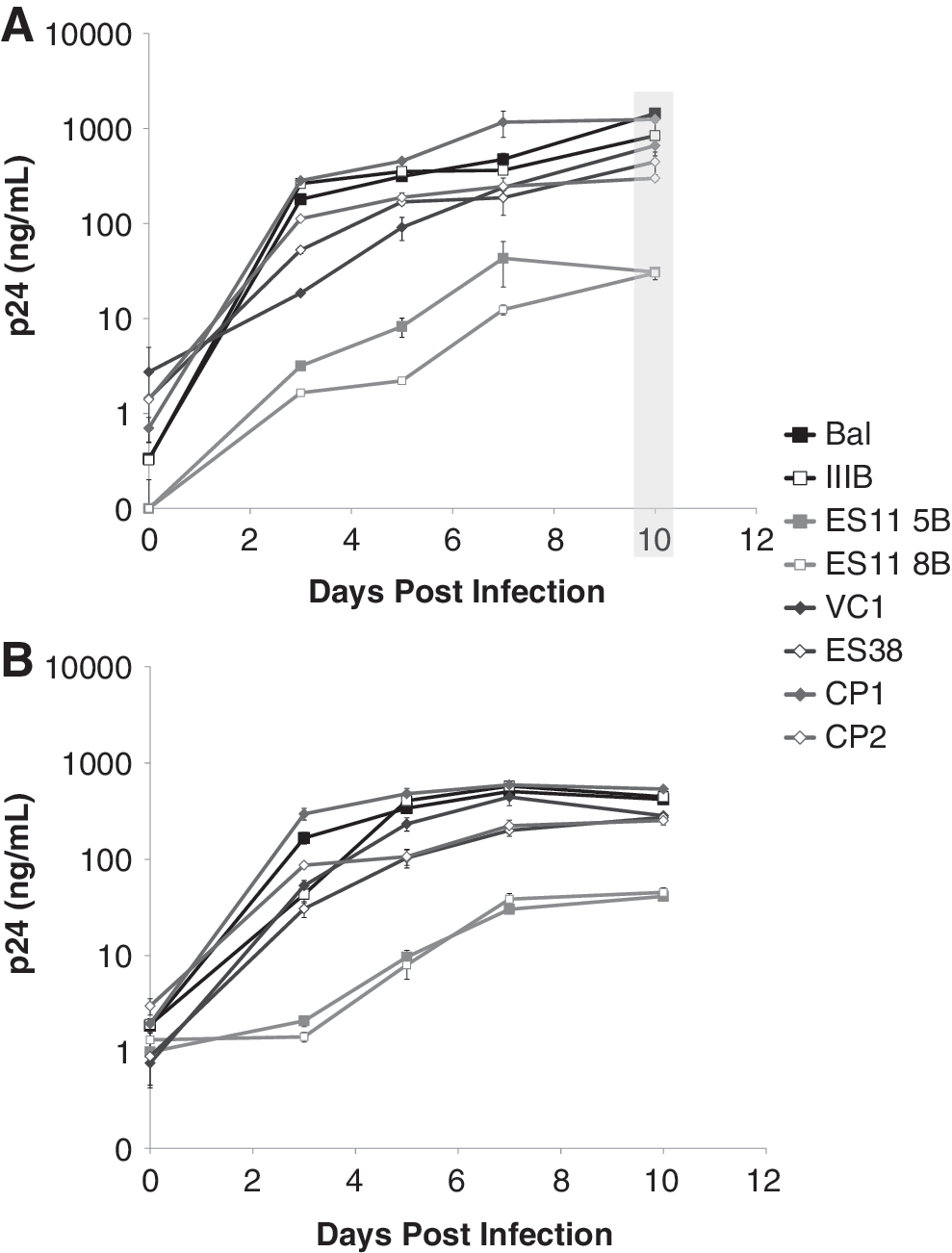
Analysis of viral fitness. Viruses from ES11 were isolated from the latent reservoir and viral fitness was analyzed over time
Full length nef sequences from virus isolated from the coculture assay, resting CD4+ T cells, and plasma virus were amplified from ES11 clonal sequences of the nef gene and were obtained by digital PCR as previously described. 11 Sequences from ES11 that were previously reported were also included in this analysis. Full genome sequences have been submitted to GenBank (accession numbers KC935957, and KC935960–KC935989). The accession numbers for previously reported sequences are EU383923–EU383964. 12
In 2006, 30 of 47 proviral clones amplified from resting CD4+ T cells contained a 38-base pair deletion resulted in a 13 amino acid deletion (52 to 64) and a frame shift mutation that resulted in a premature stop codon at amino acid position 97 (Fig. 3). The other 17 clones contained a single nucleotide compensatory deletion at position 117 that restored the reading frame and resulted in an initial K48N mutation and three downstream amino acid variations compared to consensus B clade Nef (Fig. 3). In 2007 the clones with the compensatory deletion became the dominant clone amplified from resting CD4+ T cells. Interestingly, all 7 plasma clones analyzed in 2008 contained the compensatory deletion whereas 10 out of 27 proviral clones amplified from resting CD4+ T cells contained the premature stop codon. In 2010, the majority of clones amplified from plasma and 4 of 15 proviral clones amplified from resting CD4+ T cells contained a duplication insertion that occurred after amino acid 25, as numbered using consensus B clade nef, as well as the 38-bp deletion and the compensatory single nucleotide deletion.

Evolution of the nef gene from ES11. Analysis of full genome sequencing from ES11 identified three viral variants present over time in comparison to consensus B clade nef. Partial nef sequences are shown from amino acid 0–97. One variant possessed a deletion from amino acid 52–64 (light gray highlights), in addition to a single nucleotide deletion resulting in a K48N mutation. The second variant possesses the same deletion from amino acid 52–64, but also had a duplication insertion after amino acid 25 (dark gray highlight). The third variant possessed the deletion from amino acid 52–64 but not the mutation at K48M, which results in a premature stop codon at amino acid position 97.
The 38-bp deletion was observed in the nef gene in all replication-competent isolates obtained by coculture from 2010. Some isolates also contained just the compensatory single nucleotide deletion. The fitness of a replication-competent isolate with this sequence is shown in Fig. 2 (ES11 5B). Additionally, in 12 out of 14 isolates obtained from the coculture assay, the deletion of amino acids 52 to 64 and the K48N mutation were accompanied by the duplication insertion. The fitness of a variant with this sequence is shown in Fig. 2 (ES11 8B). It thus appears that there was evolution of the nef gene over time and enrichment of isolates containing functional nef in plasma and replication-competent virus compared to virus archived in resting CD4+ T cells.
We next explored the effect of the insertions and deletions on the function of the ES11 nef isolates. We focused specifically on the ability of the Nef proteins to downregulate CD4 and MHC class I. The nef gene was amplified from proviral sequences that were representative of the most common isolates found by nef sequencing from resting CD4+ T cells and plasma virus. Two representative ES11 nef sequences were amplified in addition to NL4-3 nef. Blunt ended nef sequences were amplified and cloned into a pENTR Directional TOPO cloning vector (Life Technologies). The nef sequences were then cloned into the Vivid Colors pcDNA 6.2/EmGFP-Bsd.V5-DEST destination vector (Life Technologies). This vector is a mammalian expression vector that expresses the gene of interest, and has a separate mammalian promoter that expresses GFP. Thus, any cells that are transfected will express GFP in addition to the gene of interest.
Primary CD4+ T cells from an HLA-A*2-positive healthy donor were transfected by electroporation using an Amaxa kit and electroporator (Amaxa) with ES11 proviral nef clones and NL4-3 nef in the GFP expression vector. A no transfection control and no DNA control were used as negative controls. Cells were stained with anti-CD4 and anti-HLA-A*2 antibodies at 6, 12, 24, and 48 h after transfection. GFP-positive and GFP-negative cells were individually gated, as these cells would express patient or NL4-3 nef or have non-nef expression, respectively, and the normalized percent reduction in MFI of CD4 and HLA-A*2 expression was calculated.
The nef clone with the premature stop codon was designated ES11pr5, and the nef clone containing the compensatory one nucleotide deletion was designated ES11pr10. These clones are shown graphically in comparison to NL4-3 nef in Fig. 4A. The nef genes were isolated and inserted into a mammalian expression vector that also expressed GFP driven by an independent promoter. To study the ability of these nef genes to downregulate CD4 and MHC class I, unstimulated CD4+ T cells from HLA-A2 donors were transfected with the constructs and the GFP+ cells were gated upon for analysis. The GFP+ cells represent cells that were successfully transfected with the construct, and the percent reductions in the MFI of CD4 and HLA-A2 were calculated between the GFP+ and GFP− cells. Representative histograms for untransfected cells or cells that were transfected with NL4-3 or ES11pr10 nef clones are shown in Fig. 4B.
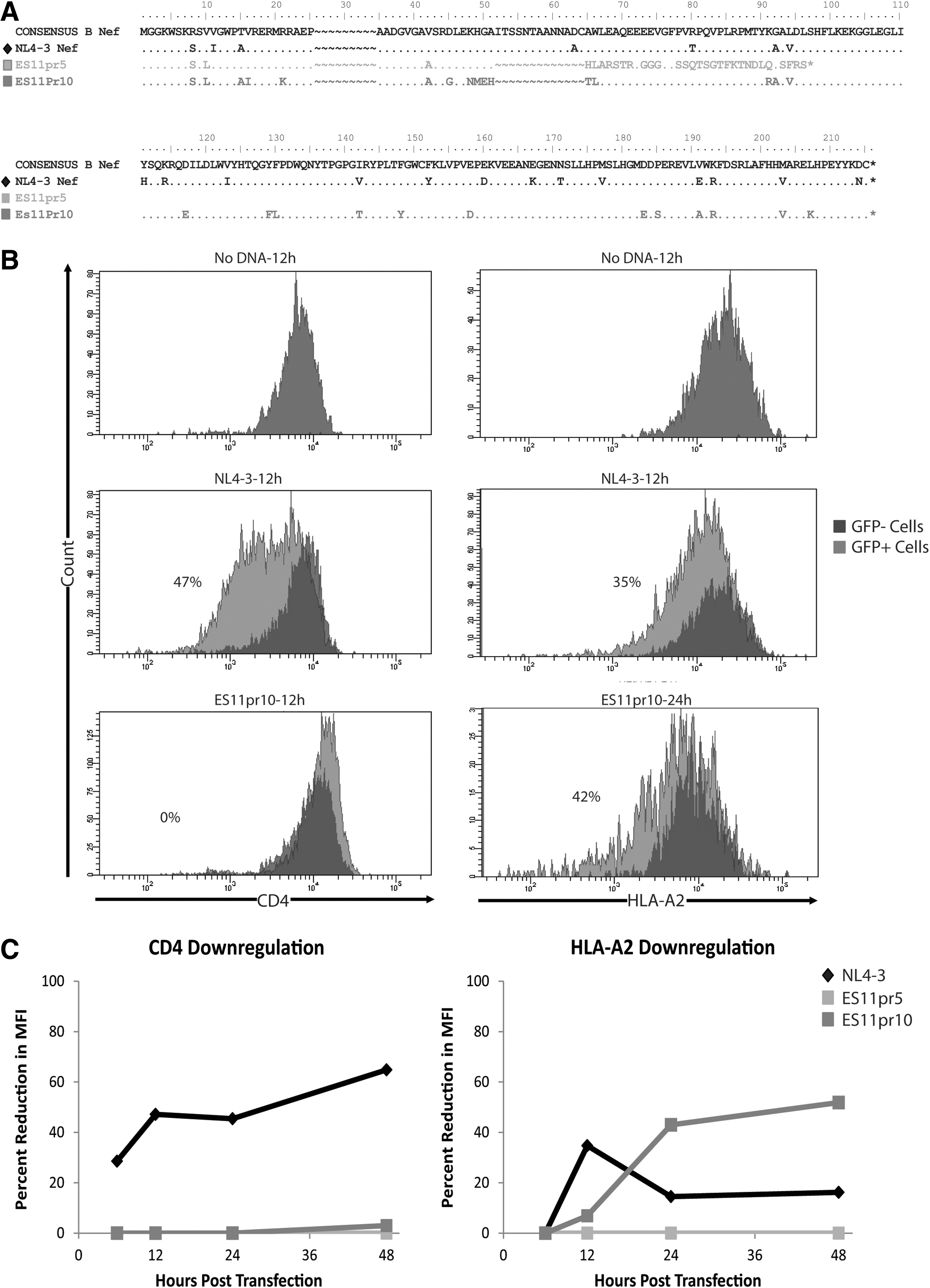
Analysis of ES11 nef function.
Nef from ES11pr5 and ES11pr10 did not mediate downregulation of CD4 at any of the time points analyzed whereas transfection with NL4-3 nef resulted in high levels of CD4 downregulation (Fig. 4C). While ES11pr5 Nef did not induce downregulation of HLA-A2 at any time point, this function was restored in ES11pr10 Nef.
Understanding the role that viral and host immune factors play in the control of viral replication is important in understanding the parameters of effective immunotherapy for HIV-1 infection. Of note, only a limited number of replication-competent viruses from ES have been characterized by full genome sequence analysis due to the difficulty in culturing virus from the latent reservoir in these individuals. Herein, we document an individual found to be infected with virus with reduced fitness, and a detailed virologic analysis was performed.
ES11 was found to be infected with a virus with a deletion in nef. Infection with a defective nef gene has been reported in some patients with long-term nonprogression and/or elite control of viral replication. 2,4,5 Nef-mediated CD4 downregulation has been implicated in reducing the rate of superinfection of HIV-1-infected cells. 13 Additionally, it has been shown that expression of CD4 decreases HIV-1 progeny virus release, due to the association of the Env protein with cell surface CD4, 14 and downregulation of CD4 has also been shown to lead to an increase in the infectivity of the progeny virus from HIV-1-infected cells. 15 Thus, the failure of the ES11 variants to downregulate CD4 could partially explain the reduction in viral fitness observed.
Interestingly, ES11 maintained undetectable viral loads for over 10 years after diagnosis with HIV-1. Only recently, has there been a detectable level of virus isolated by standard clinical assays associated with the evolution of nef and selection for viruses that did not have a premature stop codon in this gene. This evolution differs from the slow evolution due to synonymous mutations in plasma virus that we recently observed in a cohort of HLA-B*57 ES. 11 In the case of ES11, the evolution may have allowed the virus to partially evade the immune response by HLA downregulation. Thus, it appears that some HIV-1 isolates with large deletions in nef retain the ability to replicate and evolve in vivo. Interestingly, CD8+ T cells from ES11 inhibited HIV-1 replication to a similar degree as CD8+ T cells from other ES in our cohort (data not shown) and this may have prevented complete virologic breakthrough in spite of the viral evolution seen. These data confirm the importance of nef in viral pathogenesis in vivo. While many ES are infected with fully replication-competent virus, 8,9 some ES and VC may be infected with attenuated virus. A more complete understanding of the contribution of individual host and viral factors in the control of viral replication is needed for the development of effective HIV-1 vaccination strategies.
Footnotes
Acknowledgments
This work was supported by a “Sara Borrell” grant from the Spanish Health Institute (M.S.) and the NIH grant R01 AI080328 (J.N.B.).
Author Disclosure Statement
No competing financial interests exist.