
Abstract
There are few cohorts of individuals who have survived infection with HIV-1 for more than 20 years, reported and followed in the literature, and even fewer from Africa. Here we present data on a cohort of subtype C-infected individuals from rural northern Malawi. By sequencing multiple clones from long-term survivors at different time points, and using multiple genotyping approaches, we show that 5 of the 11 individuals are predicted as CXCR4 using (by ≥3/5 predictors) but only one individual is predicted as CXCR4 using by all five algorithms. Using any one genotyping approach overestimates the number of predicted CXCR4 sequences. Patterns of diversity and divergence were variable between the HIV-1 long-term survivors with some individuals showing very small amounts of variation and change, and others showing a greater amount; both patterns are consistent with what has been described in the literature.
Introduction
HIV/AIDS
People who survive for an extended period while infected with HIV-1 serve as important models for effective immunologic control of this virus, and can provide clues to natural therapeutics and therapeutic vaccines. There are few cohorts of survivors who have been followed for 20 years and those that have survived are found in Europe, America, and Australia, with the majority focused on individuals infected with subtype B. 14 –18 Very little information is available in the literature on nonprogression in Africa and even less information on subtype C [e.g., Tzitzivacos et al. 19 in children and Archary et al. 20 (in adults)] with the average follow-up being less than 2 years. Laeyendecker et al. 21 and Fang et al. 22 studied long-term survivors in Uganda and Nairobi, respectively; however, neither of these studies was based on individuals infected with subtype C. An LTS cohort in Karonga District Malawi provides a unique opportunity to investigate viral patterns in a group of primarily subtype C LTS in sub-Saharan Africa who have been followed for over 20 years. 23 –25 The extensive period of follow-up on the LTS in Karonga has allowed us to examine changes in the subtype C viral population within some individuals over a 20-year period.
In the majority of HIV-1 infections with subtype B, CCR5 tropic viruses are found to predominate during early infection. 26 –28 In approximately 50% of subtype B infections, CXCR4 usage subsequently emerges and is often associated with accelerated loss in CD4+ T cells and progression to clinical AIDS. 27, 29 –31 Studies on subtype C have reported that viral isolates almost exclusively use the CCR5 coreceptor with CXCR4 usage being observed only very rarely even within individuals with more advanced disease progression and who have advanced to AIDS. Despite the fact that subtype C represents over 50% of HIV-1 infections worldwide, relatively few CXCR4-utilizing subtype C viruses had been isolated up to 2008. 32 –39 More recently, two studies found that approximately 30% of subtype C viral isolates retrieved from individuals with advanced disease could efficiently utilize CXCR4 in vitro. 40,41 Here, we used a genotyping approach to investigate coreceptor usage among the LTS in Karonga over multiple time periods, to identify any switch in coreceptor usage as some individuals began to exhibit signs of progression.
Materials and Methods
Patients and samples
LTS were visited four times between the 1980s and 2010. Of nearly 200 individuals identified as HIV positive during population surveys in Karonga District Malawi in the 1980s, 38 were alive and agreed to give a blood sample in the 1990s. In 2004, 17 were reported to be still alive but not all individuals were seen or consented to the study and therefore dried blood spot samples were collected only from 10. In 2010 13 individuals were sought: two had died, one had left, and one refused to participate, so nine whole blood samples were collected (Table 1). Two individuals (LTS2 and LTS8) were seen in unrelated studies in 2008 and whole blood cell pellets were available from that time also. LTS21 and LTS1 were suspected to be dual infected. 23 By 2010 five LTS had been placed on antiretroviral therapy (ART) (LTS2, 5, 8, 12, and 20) and two other individuals, LTS10 and LTS21 had been referred for ART due to low CD4+ counts of 138 and 36 cells/mm3, respectively. Two LTS had CD4+ counts greater than 200 cells/mm3 (LTS1 subtype C and LTS30 unclassifiable) and have not been referred for ART (following national guidelines) (Table 1).
A different combination of three genotypic prediction tools predicted the X4?; only for LTS 2 were all five tools concordant.
LTS, long-term survivor; ART, antiretroviral therapy.
DNA extraction, PCR, and sequencing
Proviral DNA was extracted from the dried blood spot (DBS) using a QIAamp DNA Micro Kit (Qiagen) or from 200 μl of cell pellets using a QIAamp DNA Blood Mini Kit (Qiagen). Nested polymerase chain reaction (PCR) of a 750-bp region of gag p17p24 and a 549-bp region of env C2V3 was carried out as previously described. 42 A consensus sequence was produced from gel-purified PCR products where possible and for some samples more than one consensus sequence was produced. Three secondary PCR products were pooled and TA cloned using the pCR2.1-TOPO Cloning Kit (Invitrogen) or using the StrataClone PCR Cloning Kit (Agilent Technologies). Approximately 20 individual clones were sequenced in one direction. Sequence chromatographs were examined for quality in Seqman 8.0.2 (DNASTAR). All clonal data were aligned to consensus sequences already available from the 1980s, 1990s, 2004, and 2010 23,42 in MacClade 4.0 (Sinauer Associates) and additional sequences randomly chosen from the overall Karonga dataset (20 sequences retrieved in 1990s and 20 retrieved in 2008).
Sequence analysis
Phylogenetic trees were reconstructed under the GTR+gamma model of DNA substitution implemented by RAxML 7.0.3 43 with all parameters optimized by RAxML. Confidence levels in the groupings in the phylogeny were assessed using 1,000 bootstrap replicates as part of the RAxML phylogeny reconstruction. The subtype C ancestral sequences derived by Travers et al. 44 were employed as out-groups for env and gag gene trees. In addition, multiple alignments of each gene region were assembled for each LTS individually and phylogenetic trees were reconstructed in a manner similar to that described above. Pairwise evolutionary genetic distances from nucleotide sequences were computed by PAUP* 4.0 (D.L. Swofford, Sinauer Associates, Inc.) under Kimura's two-parameter model of evolution. 45 Intrapatient genetic divergence was examined by estimating the genetic distance from the earliest sequence available and all subsequent time points. Intrapatient genetic diversity at each available time point was also estimated. Coreceptor usage was determined for all env sequences for all of the LTS using five genotypic predictor tools: C-PSSM, Geno2pheno (5% FPR), CoRSeqV3-C, GPGQ, and Raymond. 41,46 –50
Results
Prediction of coreceptor tropism
LTS30, an unclassifiable subtype, was predicted as being R5 tropic. For subtype C sequences there was little congruence (15%) between all five genotypic prediction approaches. CoRSeqV3-C was found to overpredict CXCR4 usage suggesting that 94% of the subtype C sequences here were X4 tropic. When this predictor was excluded, 286 R5 and 41X4 tropic sequences are predicted as such by all four remaining methods, a concordance of 80%. CPSSM also predicted a higher number of X4 tropic sequences compared to Geno2pheno, GPGQ, or Raymond methods. All env sequences from five of the subtype C LTS (LTS1, 8, 9, 12, and 20) were predicted to be R5 tropic at all time points for which sequence information was available (Table 1 and Supplementary Table S1; Supplementary Data are available online at
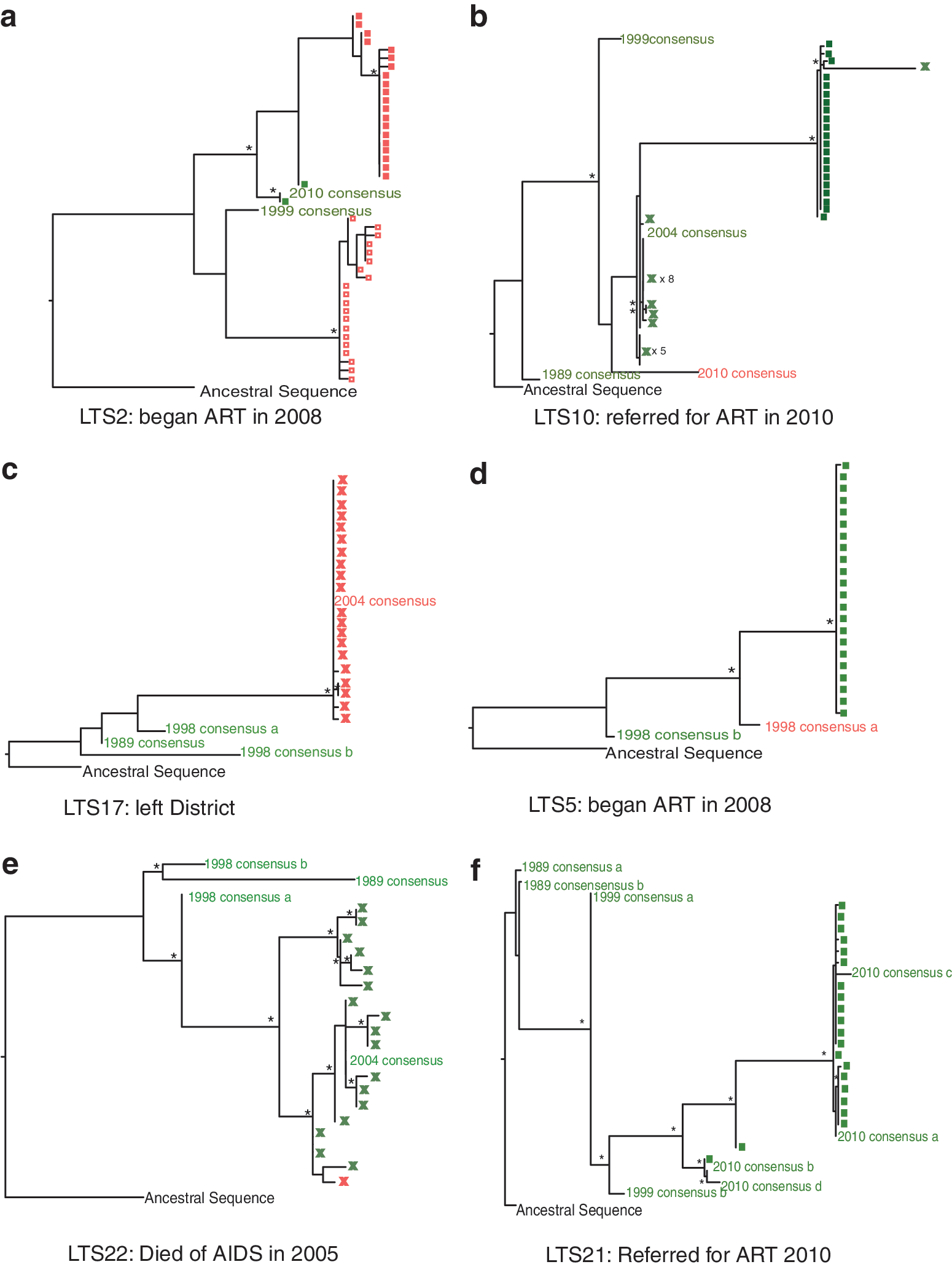
Individual maximum likelihood trees generated from all env sequences for individual long-term survivors (LTS);
The consensus sequences for LTS2 from 2010 and two of the cloned sequences from the same time point were predicted to use CCR5. The remaining clones from this time point were predicted to use CXCR4. The phylogenetic reconstruction (Fig. 1a) of the env C2-V3 fragment from LTS2 showed two viral lineages; one contained all CXCR4 tropic viruses from 2008 with the R5 consensus from 1999 as a distant sister, and the other showed a clade of CXCR4 sequences from 2010 having emerged from a CCR5 ancestral strain. LTS2 had been on ART since 2009.
One of the env V3 consensus sequences retrieved from 1999 from LTS10 was predicted to be CXCR4 tropic by CPSSM-C and CoRSeqV3-C approaches as were all sequences from 2004 with the exception of one clone retrieved from the 2004 sample. This single 2004 CCR5 sequence shared a most recent common ancestor with clones from 2010 predicted as X4 by CPSSM only. The consensus sequence from 2010 was predicted as X4 also by geno2pheno and is the only sequence color coded as X4 tropic on Fig. 1b. LTS10 had a CD4+ count of 138 cells/mm3 in 2010 and was subsequently referred for ART.
V3 sequences obtained from LTS17 in 1998 were predicted to be CCR5 tropic (Fig. 1c) but by 2004 both the consensus sequence and 20 cloned sequences were predicted to use CXCR4 by CPSSM, CoRSeqV3-C, and the Raymond method. No further follow-up information was available for LTS17, as the individual had left the Karonga region by 2010.
For LTS5 two consensus sequences were available from 1998. Of these one sequence was predicted to use CCR5 while another was predicted to use CXCR4 (by CPSSM-C, CoRSeqV3-C, and GPGQ). Cloned sequences dating from 2010 were CCR5 tropic. V3 sequence data were available from LTS22 from 1989, 1998, and 2004 (Fig. 1e); all were predicted to utilize CCR5 except one of 19 clones from 2004 (X4 by Geno2Pheno, Raymond, and CPSSM-C approaches). The patient died of AIDS in 2005.
Genetic diversity and divergence
Clonal sequences from within a time point from four of the nine LTS (LTS2, 5, 9, and 20) were homogenous (i.e., very similar if not identical) in both genes, and the consensus sequence from each time period grouped with the clones from that time period. The average gag pairwise genetic distances between all the clones from one time point within these individuals varied from 0% in LTS5 to 0.4% in LTS9 and 0.1–0.2% between all the clonal sequences from one time point in env (Tables 2 and 3). In other LTS (LTS17, 12, and 22) genetic diversity was more variable but sequences within a time period still grouped together. For LTS17 the genetic distance between gag clones was 1.6% while the env clones were homogeneous, while in LTS12 gag clones in each time period, and the env clones in 2010, were homogeneous but there was a good deal of diversity in the env clones in 2004. LTS22 contained a number of variant clones in 2004 for both genes with an average pairwise genetic distance of 1.6% in gag and 2% in env (Fig. 1, Fig. 2, Tables 2 and 3).

Individual maximum likelihood trees generated from all gag sequences for individual long-term survivors (LTS);
The two genetic divergences for LTS8, 17, 20, and 22 in the 1990s represent two different consensus sequences for that time point. The two genetic divergences for LTS5 in 2010 represent two consensus sequences available from the 1990s, which were both used to calculate the amount of genetic divergence by 2010.
Within LTS8, by comparison, gag and env clones did not always group with the consensus sequence of the same time period (e.g., Fig. 2d). Within LTS10 the gag clones from 2010 exhibited a high level of genetic diversity and the 1999, 2004, and 2010 consensus sequences (Fig. 2e) all grouped within the same clade as these clones. The clones from 2004 formed a separate but homogeneous sister clade to this with both lineages supported by high bootstrap values. However, the env 2004 and 2010 cloned sequences from LTS10 (Fig. 1b) showed very little variation within each time point with the exception of one clone from 2004 that grouped within the clones from 2010. A large amount of change had occurred between 2004 and 2010 resulting in a long branch separating the two clades with an average pairwise genetic distance of 12.2% between the two sets of clones. Seven of the nine individuals included showed an overall increase in pairwise genetic divergence between the first sampling time point in 1988/89 and the last sampling time point of either 2004 (LTS9, 17, and 22) or 2010 (LTS5, 8, 9, and 10) (Tables 2 and 3). However, in LTS12 a decrease in divergence was seen between the first sequence information from 1999 and the last sampling time point in 2010 in both genes (Tables 2 and 3).
Discussion
Early studies of subtype C tropism reported little if any usage of the CXCR4 coreceptor. 32,34,39,51 More recently, Connell et al. 40 found that 30% of isolates from 20 subtype C-infected individuals with AIDS from South Africa were able to use CXCR4 for cell entry. Raymond et al. 41 detected CXCR4 tropic virus in 29% of 52 subtype C-infected individuals who had started ART in Malawi, while Jakobsen et al. 52 describe CXCR4 tropic virus detected in one of 21 ART-naive individuals in Zimbabwe who had progressed to advanced stages of HIV-1subtype C infection. In this study while CXCR4 tropic viruses were predicted in 5 out of 11 individuals by multiple genotypic prediction tools, in only one individual was X4 usage predicted by all five approaches.
Studies on subtype C samples where both phenotypic methods and C-PSSM were used showed good concordance between the results (C-PSSM displayed a specificity of over 93% in detecting CXCR4 tropism). 40,41,47 In addition, the European Consensus Group on clinical management of HIV-1 tropism testing has recently indicated that genotyping approaches can be sufficient for determining resistance to CCR5 antagonists. 53 Due to a lack of appropriate samples at each time point phenotypic approaches could not be pursued for these individuals. This work clearly shows, however, that additional work needs to be done to elucidate the relationship between these two approaches for successful development of a genotypic predictor tool.
LTS10 was referred for ART in 2010 as the CD4+ cell count had dropped to 138 cells/mm3 indicating progression, which was 6 years after viruses able to use CXCR4 are predicted (but only by two genotypic predictor tools). This may suggest that a switch to CXCR4 tropism within subtype C is not as strongly correlated with disease progression, consistent with the suggestion by Meehan et al. 54 that CXCR4 usage can be transitory during disease progression. It may be that genotypic prediction by some but not all predictor tools indicates an intermediate phase or a dual phase for coreceptor usage or indeed that genotypic predictor tools are not yet sensitive enough to accurately predict coreceptor usage in subtype C. Of the six LTS who have been placed on ART, suggesting disease progression, four were CCR5 tropic, and of the two who had died one (LTS9) was at no time predicted to utilize CXCR4. However, as all of these individuals were classified as LTS, further exploration of the emergence of CXCR4 usage in nonprogressors as well as normal progressors is required to fully understand the implications within subtype C-infected individuals.
Of the nine individuals infected with subtype C, who were not suspected to be dual infected, clonal sequences generated of both env and gag from four (LTS2, 5, 9, and 20) of the LTS appeared highly homogeneous with a diversity of below 1.5% in env and 0.5% for gag. There are no data available for diversity patterns within LTS; however, low levels of viral diversity have previously been recorded within LTNPS. 55 –58 Bello et al. 56 reported a mean heterogeneity in env of <1% in seven LTNPS who were in year 8 to 15 of infection. Braibant et al. 58 noted that viral diversity did not exceed 1% in three LTNPS who had been HIV positive for over 8 years with stable CD4+ cell counts. Low levels of diversity have been suggested to be indicative of variants of lower fitness, which in turn has been associated with nonprogression. 59,60 However, the four individuals studied here who apparently harbor viral populations of extremely limited diversity have all shown signs of progression, i.e., low CD4+ cell counts and placement on ART, so the low diversity detected may instead be a result of homogenization of the viral population associated with advanced disease.
It is also possible that the PCR and cloning approach may have contributed to the limited viral diversity in some individuals included in this study. This method may be influenced by reduced template detection in the early stages of PCR. Currently it is impossible to tell to what extent experimental design may have impacted the results. Single genome analysis has been cited as a more efficient method in the detection of viral diversity. 60 However, Jordan et al. 61 comparing standard PCR/cloning to single genome sequencing determined that both methods are likely to provide a similar measure of viral population diversity within a given sample. Clonal sequences generated for LTS5 were almost identical in env and gag, indicating a possible lack of viral evolution and restricted viral diversity rather than PCR bottlenecking. Twenty identical clonal sequences were identified in an LTNPS 9 years after infection in a study by Bello et al. 56 with a second LTNPS producing 39 almost identical clones 17 years after infection in the same study. In LTS8, the high levels of viral diversity in env in 2008 (4%) were followed by a sharp decline in diversity in 2010 (0.6%) with identical clonal sequences generated. This restricted clonal diversity in LTS8 2010 was not mirrored in gag clonal sequences (1.2`%), which points to a possible PCR bottlenecking in the 2010 env sample.
Higher levels of genetic diversity in env and gag were seen in LTS10 and 22, and in env for LTS12 and gag for LTS17. Across these three individuals the intraindividual genetic distance in gag ranged from 0.6% to 2.6%, and in env ranged from 1.8% to 4%, which are also comparable to diversity values described in the literature. Very few viral diversity studies have focused on gag. Huang et al. 12 described an average of 1.7% diversity in full gag (range 0.03–3%) in eight LTNPS 12–15 years after infection, while Braibant et al. 58 described a range of viral diversity from <1% to 5.6% in env V1-V5 in nine LTNPs, both similar to what was recorded here.
In normal progressors, as HIV-1 infection progresses, the virus accumulates new mutations resulting in an increase in genetic divergence over time from the original infecting strain. This increase in divergence was also observed here in eight of the LTS from Karonga (LTS2, 5, 8, 9, 10, 17, 20, and 22) for whom data are available. The observed increase in divergence within these individuals indicates that evolution and replication have not been arrested in spite of the observed slow disease progression. Different rates of divergence were seen between the LTS and may be the result of the different rates of evolution of the HIV-1 virus within each individual, making it impossible to identify a pattern unique to LTS. Within two individuals (LTS12 and LTS20) a decrease in viral divergence was seen after an initial increase in divergence in 2004. LTS12 began ART in 2005 and LTS20 began ART in 2006, thus altering the environment within which the HIV-1 virus replicates and evolves. This decrease in divergence, however, may also be due to the sampling of an ancestral lineage that had existed previously as a minor variant and had remained latent, as has been described in other individuals. A similar pattern was shown by Bello et al. 56 where 7 of 16 LTNPs were seen to show slow or arrested viral divergence within the C2-V5 env region in conjunction with reduced viral diversity and viral loads.
Out of the 12 LTS studied, two individuals (LTS1 and LTS21) were identified as having possible dual infections. 23 LTS1 and LTS21 are of specific interest because both of them are still therapy free after 21 years (although LTS21 had a CD4 count of <200 cells/mm3 in 2010, Table 1) and both were identified as putative dually infected individuals via phylogenetic analyses of consensus sequences. 23 Braibant et al. 58 reported that in a cohort of nine LTNPs three individuals, i.e., 30% (n=9) were identified as being either coinfected or superinfected with two different strains of HIV-1. Bello et al. 56 reported that one individual within a cohort of 15 LTNPs, i.e., 6.25% (n=15) was infected with two separate viral strains. The high level of superinfection within nonprogressors may be due to the extended period of infection, increasing the possibility of reinfection occurring. Phylogenetic reconstruction of all sequences (consensus and clonal) from each LTS and 40 control sequences, for both env and gag, showed all sequences from each individual formed monophyletic clusters with the exception of these two LTS (data not shown). Further analyses (SH tests) suggests that one of the LTS (LTS21) is likely to be dually infected, but data from the other (LTS1) are also consistent with the sampling of a single diverse population. Both are females; the former individual (LTS21) was 31 in 1999 and had already had three children from two fathers. The latter individual was older (born circa 1950), had one husband, and had a low risk of superinfection. Ultradeep pyrosequencing is likely to be the best way forward for detecting multiple infections.
Studies on viral diversity of long-term survivors and nonprogressors are often based on more regular sampling intervals but over shorter periods of time than we have presented here. Many are focused on small subsets of patients using different gene regions, sampling times, frequency of sampling, and number of sequences, making it very difficult to draw any direct comparisons or detect patterns between the data described here and that described elsewhere. However, this work is consistent with both types of patterns described in the literature for diversity within LTS, i.e., some individuals showing very reduced diversity, with others showing a greater amount of diversity. 55 –58 The inability to differentiate between methodological bottlenecking and detection of highly homogeneous populations makes it difficult to draw definitive conclusions from diversity data. Only studies employing ultradeep sequencing with the primer ID approach will determine the extent to which both the cloning and single genome amplification approaches have affected measures of diversity within hosts. This study, however, suggests a much higher proportion of CXCR4 variants within subtype C-infected individuals than previously thought, and at an earlier stage of infection. Additional unpublished data from the general population in this district also suggest a higher proportion of CXCR4-using virus than expected (Seager et al., unpublished data). These observations may have implications for treatment regimes in the future in this cohort.
Sequence Data
All sequences have been deposited into GenBank; accession numbers (KJ738309–KJ738330, KJ809622–KJ809918, and KJ809919–KJ810193) and alignments are available from the corresponding author.
Footnotes
Acknowledgments
This material is based upon works supported by Science Foundation Ireland under grant 07/RFP/EEEOBF424 and by an IRCSET EMBARK Scholarship to Ishla Seager (RS/2007/203). The Karonga Prevention Study is funded primarily by the Wellcome Trust, with contributions from LEPRA. Permission for the study was received from the National Health Sciences Research Committee, Malawi, and the Ethics Committee of the London School of Hygiene and Tropical Medicine, UK.
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.