
Abstract

E
We now report on the in vivo anti-HIV activity of IM in a preclinical mouse model, NOD/SCID/IL-2Rγnull (NSG) mice engrafted with human peripheral blood lymphocytes (PBLs) and infected with HIV. 16 Human PBL-engrafted mice were intraperitoneally (ip) injected with 20,000 units of 50% tissue culture infective dose (TCID50) of HIV, followed by daily ip treatment with IM (10–40 mg/kg/day, dissolved in 10% Cremophor EL) or phosphate-buffered saline (PBS) (in 10% Cremophor EL solution) for 18 days. IM was purchased from Cayman Chemicals (Ann Arbor, MI) and Cremophor EL from Sigma (St. Louis, MO). IM doses were based on previous reports demonstrating lack of toxicity. 17 –19 Blood samples, drawn from the retroorbital vein on days 9 and 18 after infection, were analyzed for plasma HIV RNA copy number (quantitative RT-PCR) and human CD4/CD8 ratios (flow cytometry analysis). Nonparametric statistical analyses of the data were performed using the Mann–Whitney U test (GraphPad Prism Software, La Jolla, CA); p<0.05 was considered significant.
In a first experiment, we evaluated the in vivo activity of IM against multidrug-resistant molecular clone NL437303-3, which carries the reverse transcriptase (RT) gene amplified from plasma of a patient with multidrug-resistant HIV. 20 This virus has mutations 41L, 67N, 210W, 215Y, 69D, 44D, and 118I, conferring resistance to thymidine analogs (AZT, d4T), guanosine analog (ABC), and adenosine analogs (ddI and TDF). On day 9, HIV RNA was undetectable in most samples (not shown). Day 18 data are shown in Fig. 1a. In mice given PBS (n=6), mean plasma HIV RNA (copies/ml) was 7.6×106 (range, 2.5×106 to 7×107). In IM-treated mice, mean HIV RNA (copies/ml) was reduced to 1.7×106 (range, 3.4×105 to 1×107; n=5; p=0.17), 2.3×105 (range, 1.1×104 to 1×106; n=6; p=0.002), and 3.8×104 (range, 1×104 to 1.3×105; n=5; p=0.004) at 10, 20, and 40 mg/kg/day doses, respectively. The mean CD4/CD8 cell ratio in mice given PBS was 0.021 (range, 0.01 to 0.03). In contrast, the ratios were significantly higher in IM-treated mice. The mean CD4/CD8 ratios were 0.24 (range, 0.14 to 0.34; p=0.007), 0.34 (range, 0.16 to 0.55; p=0.004), and 0.19 (range, 0.07 to 0.32; p=0.007) at the 10, 20, and 40 mg/kg doses, respectively. IM did not significantly impact body weights in treated animals (p>0.1 in all three IM treatments compared to PBS treatment) or have any apparent toxicity. Together, these data demonstrate that IM reduces multidrug-resistant HIV NL437303-3 replication in vivo.
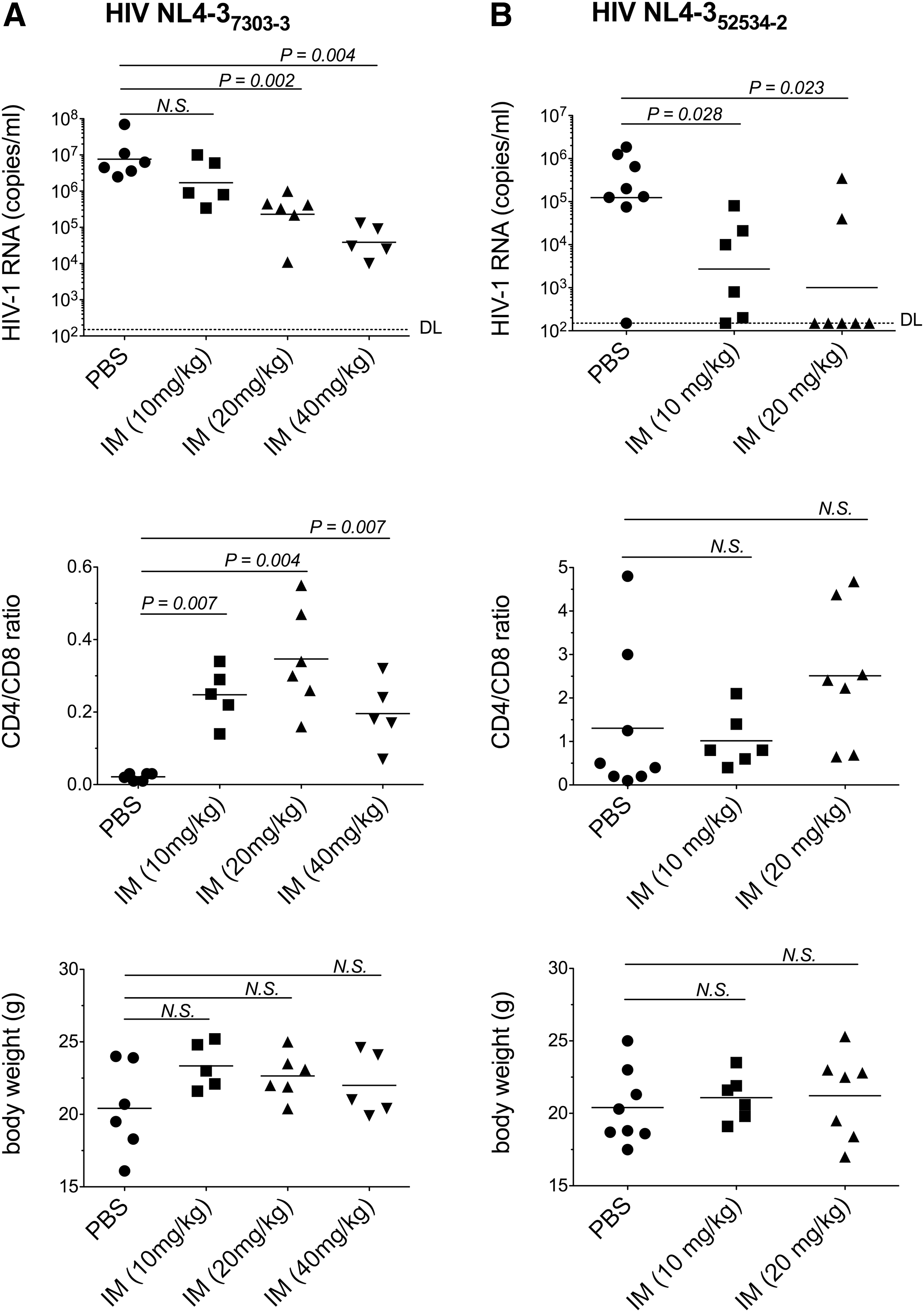
Indirubin-3′-monoxime (IM) reduces replication of multidrug-resistant HIV in humanized mice. Six- to 8-week-old female mice were intraperitoneally (ip) injected with peripheral blood lymphocytes (PBLs) (107 per mouse) from healthy donors. Three weeks later, successfully engrafted mice were ip injected with 20,000 TCID50s of HIV molecular clones NL4-37303-3
We confirmed the in vivo activity of IM using another virus, multidrug-resistant molecular clone NL4352534-2 (Fig. 1b). NL4352534-2, also derived from a patient with multidrug resistance, carries mutations 41L, 210W, 215Y, 184V, 69 insertion, and 74V. 20 The presence of the 69 insertion and the 184V mutation, together with thymidine analog mutations, confers high-level resistance to all nucleoside reverse transcriptase inhibitors (NRTIs), including the cytosine analogs 3TC and FTC. 21 IM was used at doses of 10 and 20 mg/kg/day. As with NL437303-3, plasma NL4352534-2 RNA levels were undetectable on day 9 (not shown). On day 18, PBS-treated mice (n=8) had a mean HIV RNA (copies/ml) of 1.2×105 (range, ≤150 to 1.8×106). IM significantly suppressed HIV RNA to a mean of 2.7×103 (range, 150 to 8×104; n=6; p=0.028) at 10 mg/kg, and to a mean of 1×103 (range, 150 to 3.5×105; n=7; p=0.023) at 20 mg/kg. Mean CD4/CD8 ratios were 1.3 (range, 0.10 to 4.80) in PBS-treated mice, 1.01 (range, 0.40 to 2.1; p=0.4) at 10 mg IM/kg, and 2.51 (range, 0.65 to 4.68; p=0.11) at 20 mg IM/kg. Again, IM treatment did not impact body weight or have any identifiable adverse effects (not shown).
Together, the data demonstrate that IM suppresses viremia of two reference multidrug-resistant HIV molecular clones in a preclinical animal model. Although IM reduced plasma viremia by more than 2 log10 units in both virus infections, HIV RNA levels were still detectable and relatively high in some of the treated animals. This suggests that treatment with one drug alone may not be sufficient to completely inhibit plasma viremia in this animal model. In agreement, single treatment with EFdA (a potent NRTI in clinical development) decreased plasma viremia by ∼2 log10 units, yet to detectable levels, in a similar mouse model. 22 Other limitations of the animal model used are the absence of multilineage hematopoiesis and of primary immune responses. 23 Thus, any potential contribution of IM to HIV treatment in patients should be viewed in the context of combined ART and of a whole human immune system.
Consistent with reductions in viremia and decreased CD4+ T cell depletion, NL437303-3-infected mice treated with IM had significantly higher CD4/CD8 ratios than did PBS-treated controls. Although we did not measure CD4/CD8 ratios in uninfected mice, ratios ranging between ∼0.5 and 2 have been reported by others in uninfected NSG mice. 16,22,24 In NL4352534-2-infected mice, IM caused a significant change in viremia but not in CD4/CD8 ratios. That the CD4/CD8 ratio did not change in infection with NL4352534-2 might be due to low virus replication levels (mean HIV RNA copies/ml of 1.2×105 in PBS-treated NL4352534-2 versus 7.6×107 in PBS-treated NL437303-3 mice) and thereby reduced CD4+ T cell killing, consistent with the reduced viral fitness associated with the 184V mutation in NL4352534-2. 25
In clinical trials of indirubin (parent compound of IM) in China, daily oral doses of 150–450 mg were effective against chronic myelogenous leukemia. 13 Treatment was well tolerated and the main side effect was gastrointestinal tract problems. However, indirubin pharmacokinetic data were not reported. Currently there are only limited animal data on the toxicity of IM, 17 –19 and no validated human or animal data on pharmacokinetics, making it difficult to predict what doses will inhibit HIV in patients. In our experiments, IM was administered via ip to reduce variability often observed with oral administration. Since oral indirubin is effective against leukemia and IM is more soluble than indirubin, we expect that oral administration of IM will be effective against HIV in patients in the absence of toxicity. In future studies we plan to evaluate the pharmacokinetics and pharmacodynamics of IM. We will also evaluate toxicity by examining blood chemistry profiles and blood counts as well as toxic lesions in different organs, both in IM treatment alone and in combination with ARTs.
In summary, these data demonstrate inhibition of multidrug-resistant HIV by targeting viral gene expression, a step of the viral cycle not targeted by current ARTs, with IM in vivo. Despite the limitations of the animal model, 23 IM, and perhaps flavopiridol analogs 26 and recently developed mimetic CDK9 inhibitors, 27 may provide effective approaches for controlling HIV, particularly drug-resistant strains, in patients.
Footnotes
Acknowledgments
This work was supported by research funds from the Institute of Human Virology.
Authors contributions: A.H., S.N., M.R., J.B., and R.R.R. designed the experiments; A.H., S.N., S.M., J.Z., and N.M.L. performed the experiments; A.H., M.R., J.B., and R.R.R. contributed to data analysis, discussion of results, and writing of the article. We thank Lanea George (Animal Facility, IHV) for outstanding technical assistance. We thank George L. Drusano (Institute of Therapeutic Innovation, College of Medicine, University of Florida) for critical reading of the article and input on future pharmacokinetic studies.
Animal protocols were approved by the Institutional Animal Care and Use Committee, University of Maryland School of Medicine.
HIV-1 molecular clones carrying RT sequences amplified from the plasma of patients with multidrug resistance were obtained from Dr. Robert Shafer (Stanford University School of Medicine, Stanford, CA) through the NIH AIDS Repository (Gaithersburg, MD).
Author Disclosure Statement
No competing financial interests exist.