
Abstract
HIV infection is a risk factor for the tumorigenesis including non-AIDS-defining cancers such as those of the gastrointestinal tract. However, the mechanisms underlying such cancer outgrowth are still unknown. Furthermore, combined HIV/cancer studies are difficult to evaluate using primate models or in the clinical patient setting. To understand the mechanisms of tumor outgrowth in the context of HIV infection, we adopted a humanized mouse model permissive to infection and cancer as well as an in vivo humanized mouse challenge with colon cancer in the context of HIV infection. Immunodeficient NOD SCID IL-2R–/– mice were immunologically reconstituted by adoptive transfer of 107 HIV-negative donor peripheral blood leukocytes and challenged with 106 HCT116 human colon cancer cells. A group of mice was treated with antiretroviral therapy. Tumor microenvironment and epithelial tissues in the context of HIV infection were analyzed using immunohistochemistry. We demonstrate that HIV-infected humanized mice develop significantly larger tumors than uninfected mice (p<0.05). Epithelial cell proliferation in HIV-infected mice is significantly enhanced in comparison to proliferation in uninfected mice (p<0.01). Moreover, the activation of β-catenin, an important step in intestinal epithelial cell proliferation and tumorigenesis, is elevated in the tumors of HIV-infected mice (p<0.0001). Importantly, antiretroviral therapy reverses these pathological processes independently of CD4+ T cell return. These findings model the ability of HIV infection to result in tumor outgrowth that is evident in HIV-positive patients and lend insight into previously unrecognized mechanisms that may underlie this pathology.
Introduction
I
In our current study, we adopted a state-of-the-art small animal [NOD SCID IL-2R–/– Hu-PBL (NSG-HuPBL)] model to evaluate the interplay between HIV and colon cancer. The NSG-HuPBL humanized mouse model has been established as a valid model for the study of human diseases, including infection and cancer. 6 We chose this model for the current studies because humanized mice represent one of the only small animal models in which HIV can in vivo infect human PBLs. 7,8 We report, here, that HIV-infected humanized mice develop significantly larger colon cancer (HCT116) tumors than mice without HIV infection. This model correlates with clinical observations that have demonstrated that colon cancer outgrowth is increased in the context of HIV infection.
Materials and Methods
NSG-HuPBL immune reconstitution and HIV infection
Six- to eight-week-old, specific-pathogen-free, male, NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ mice (Jackson Laboratories, La Jolla, CA) were housed in animal facilities designed for immunocompromised mice at Rush University Medical Center and utilized in accordance with Rush Institutional Animal Care and Use Committee (IACUC) and Institutional Biosafety Committee (IBC) guidelines. Peripheral blood from HIV-negative donors was obtained through peripheral venous blood collection in accordance with guidelines on human research and approval of the Institutional Review Board of Rush University Medical Center. Written informed consent was obtained from all donors in accordance with the Declaration of Helsinki. Peripheral blood mononuclear cells (PBMCs) were obtained by peripheral venipuncture from two HIV-negative HLA-A2+ donors. A single donor was used for humanization of all mice each time the experiment was conducted or repeated. For humanization of mice, 107 human peripheral blood mononuclear cells [in 200 μl of phosphate-buffered saline (PBS) after Ficoll-Hypaque density gradient centrifugation] were transferred via intraperitoneal (IP) injection on day 0. On day 7, the mice were infected with HIV-1Ba-l 104 TCID50/ml (tissue culture infectivity dose) by IP injection. HIV-1Ba-L is a full infectious virus obtained from the AIDS reagent and reference program (see the Acknowledgments). After 2 weeks, HIV infection was characterized by p24 expression within CD4+ T cells by flow cytometry (as described in the “Flow cytometric analysis of cells” in the Materials and Methods subsection) and HIV LTR measurement by real-time PCR, as previously described. 9
Tumor challenge
Human colon cancer HCT116 cells were mixed with a matrix gel (BD Matrigel, BD Biosciences, San Jose, CA) prior to injection at a 1:1 volume/volume ratio and kept on ice. Mice were challenged with 100 μl (106 HCT116 cells) via subcutaneous injection into the right hind flank. 10 Tumors were collected and analyzed 2 weeks posttumor challenge.
Antiretroviral treatment studies
For studies involving antiretroviral treatment, mice were infected with HIV-1Ba-l (TCID50/ml) on day 0, and treated on days 7, 9, and 11 by IP injection 11,12 of zidovudine (AZT; nucleoside analog reverse transcriptase inhibitor; 25 mg/kg weight), nelfinavir (protease inhibitor; 10 mg/kg weight), and efavirenz (nonnucleoside analog reverse transcriptase inhibitor; 25 mg/kg weight). In these experiments, mice were challenged with HCT116 tumors on day 14 and dissected on day 28.
Flow cytometric analysis of cells
Cells were obtained from spleens of mice using mechanical dissociation and resultant single-cell suspensions were stained using fluorescently labeled antibodies against cell surface markers, according to standard protocols described previously. 5,13 All antibodies were purchased from eBioscience, Inc. (San Diego, CA). Flow cytometry was performed using a BD FACS Canto II flow cytometer (BD Biosciences) and data were analyzed using FlowJo software (Tree Star, Ashland, OR). For the gating strategy, live leukocytes were identified by using a lymphocyte (FSC-A versus SSC-A) gate followed by a CD45 versus Live/Dead gate [where leukocytes are CD45+ and live cells are negative for Fixable Viability Dye (eBioscience)]. Doublets were then excluded using SSC-A versus SSC-H and FSC-A versus FSC-H gating.
HIV DNA detection
Blood cells were obtained by centrifugation and removal of plasma from approximately 200 μl of blood. Genomic DNA was isolated using the DNeasy Blood and Tissue Kit (Qiagen, Valencia, CA). Concentrations were estimated at a wavelength of A260. Approximately 50 ng/sample was used to amplify a small region of the HIV LTR (F-5′-TCAAGTGAGTGCCCGGTT and R-5′-AGCTCCGGTTTCTCTTTCGCT primers) and GAPDH (F-5′-TGACTTCAACAGCGACACCCACT and R-5′-ACCACCCTGTTGCTGTAGCCAAAT primers) by PCR using AmpliTaq Gold polymerase (Applied Biosystems) at 95°C for 10 min followed by 30 cycles of 95°C for 15 s, 60°C for 45 s, and a single cycle of 72°C for 10 min. Amplicons (2 ml) were run on a 2% agarose gel and stained with ethidium bromide to detect DNA.
Histology staining and immunostaining
Epithelial tissue and tumors were freshly isolated and paraffin embedded after fixation with 10% neutral buffered formalin. For immunohistochemistry staining (IHC), slides were processed using hematoxylin-eosin (H&E) and observed under a microscope. IHC was performed on 5-μm paraffin-embedded sections. After preparation of the slides as described previously, 14 tissue samples were incubated with anti-PCNA and anti-β-catenin antibodies (BD Transduction Laboratories). BrdU staining was performed as previously described. 14 Immunostaining was performed as previously described. 14 Anti-CD45 antibody (H-230) was purchased from Santa Cruz Biotechnology Inc. Mouse anti-CD68 antibody (ab955) is from Abcam.
Western blot analysis and antibodies
Protein expression in epithelial cells and tumor tissues was detected by Western blot. Epithelial cells were collected by cutting or scraping, as previously described. 15 Briefly, cells were lysed in lysis buffer [1% Triton X-100, 150 mmol/liter NaCl, 10 mmol/liter Tris, pH 7.4, 1 mmol/liter EDTA, 1 mmol/liter EGTA, pH 8.0, 0.2 mmol/liter sodium orthovanadate, and protease inhibitor cocktail (Roche Diagnostics, Barrington, IL)]. An equal amount of protein was separated by SDS-polyacrylamide gel electrophoresis, transferred to nitrocellulose (Bio-Rad, Hercules, CA), and immunoblotted with primary antibodies: anti-phospho-β-catenin, anti-β-catenin, or anti-β-actin (Sigma-Aldrich, Milwaukee, WI) antibodies and visualized by ECL chemiluminescence. Protein bands of interest were quantitated densitometrically and normalized to β-actin.
Statistical analysis
Data are expressed as mean±SD. All statistical tests were two-sided. p values < 0.05 were considered statistically significant. Differences between groups were analyzed using ANOVA with Dunnett's test for multiple comparisons using GraphPad Prism software version 5 (GraphPad Software, Inc., La Jolla, CA).
Results
Colon cancer outgrowth is increased in the context of HIV infection
To investigate the impact of HIV infection on tumor outgrowth in vivo, we adopted an NSG-HuPBL cancer xenograft mouse model 16 using HIV infection and human colon cancer (HCT116) cell challenge. 10 To enable HIV infection within this model, 107 human PBLs from healthy donors were adoptively transferred into NSG mice on day −7 via IP injection. On day 0, a group of PBL-reconstituted NSG mice was infected with HIVBal (104 PFU) via IP injection. In our model, by day 14 (2 weeks after HIV infection) CD4+ T cells were dramatically depleted in spleens of HIV-infected mice in comparison to control mice (Fig. 1a) and both HIV p24 expression and HIV DNA long terminal repeat (LTR) were detected in HIV-infected mice (Fig. 1b and c). No significant physiological changes in body weight and liver size were noted (data not shown). Toward understanding how such HIV infection impacts tumor outgrowth, HIV-infected and HIV-uninfected mice were challenged with HCT116 colon cancer cells (106) 1 day after HIV infection and tumors were harvested 2 weeks postchallenge (Fig. 2a). Tumors were significantly larger and heavier in HIV-infected mice in comparison to HIV-uninfected mice (p<0.05) (Fig. 2b–d) despite the fact that the mouse body weights were similar (data not shown).
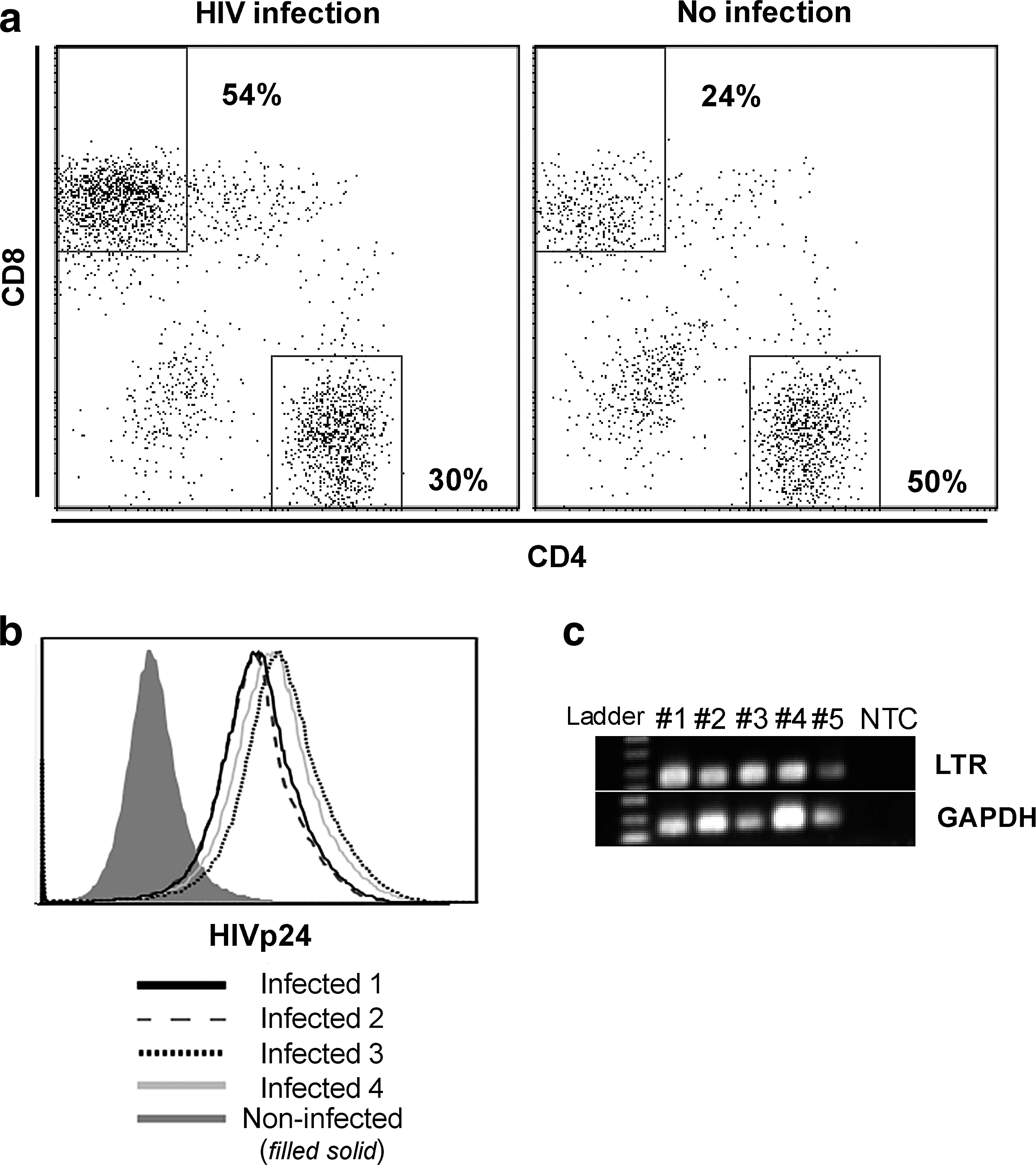
HIV infection in the NSG-HuPBL mouse model.

HCT116 colon cancer outgrowth in the context of HIV infection.
Changes in tumor cell proliferation and β-catenin expression in the context of HIV infection
Toward elucidating the changes that take place within colon cancer cells in the context of HIV infection that may have led to the increased tumor outgrowth that we observed in Fig. 2, we analyzed tumor tissue by H&E. Such staining showed increased pathological changes involving tumor tissues from HIV-infected mice compared with noninfected mice, including inflammation surrounding the tumor, necrosis, and mitosis (data not shown). IHC of PCNA, a marker for cell proliferation, was significantly enhanced in the tumor tissue of HIV-infected mice compared to that of uninfected mice (p<0.01; Fig. 3a and b, PCNA) despite the similar immune cell infiltration of the tumor (indicated by CD45 staining) seen in HIV-negative versus HIV-positive groups (Fig. 3c). In HIV-negative versus HIV-positive groups, the macrophage marker CD68 did not show a significant difference either (Fig. 3c, CD68+ cells are red).
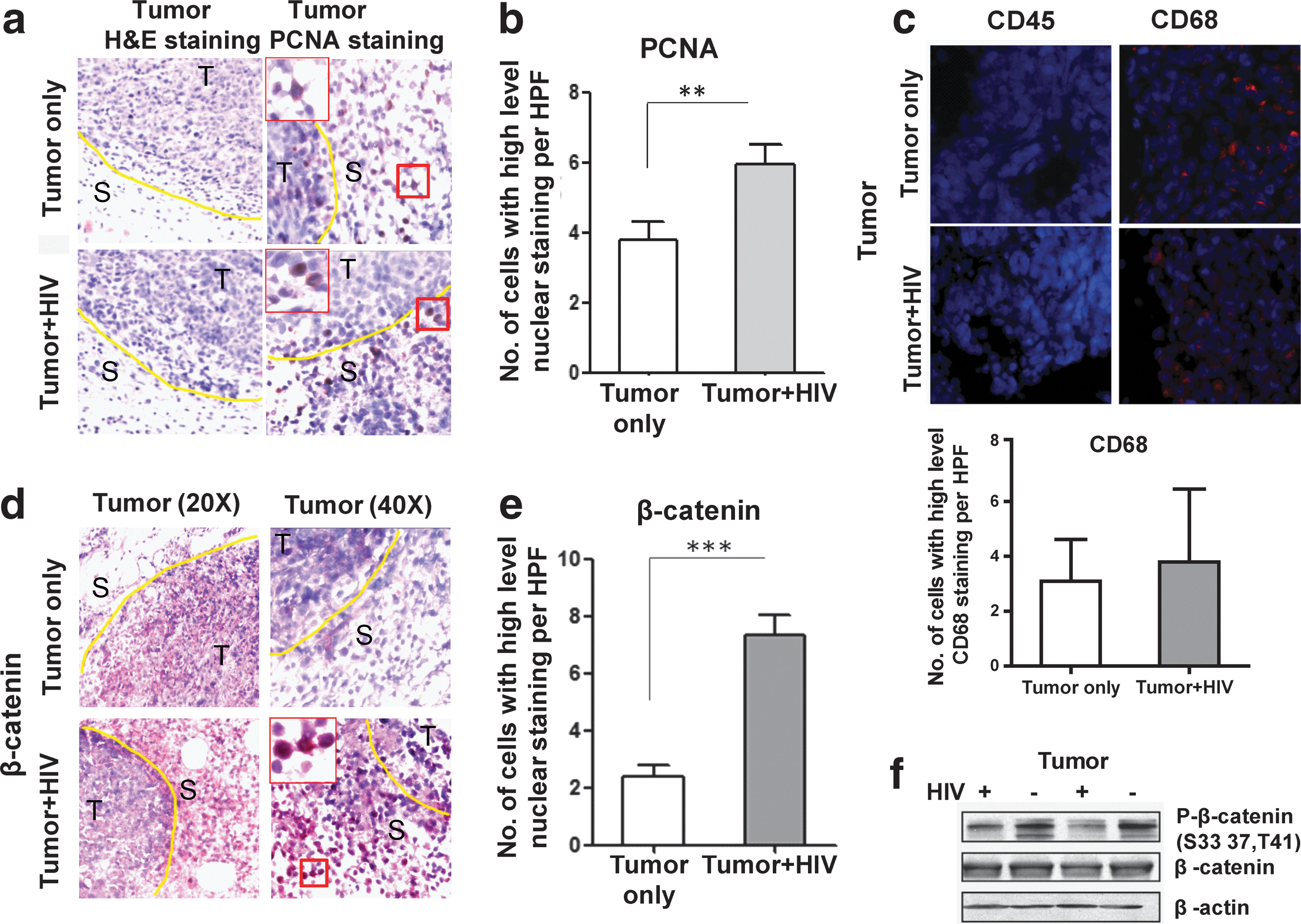
Pathological and β-catenin expression changes in the tumors of HIV-infected NSG-HuPBL mice.
Since activation of the β-catenin pathway is associated with the initiation of intestinal neoplasia, we evaluated changes in the β-catenin signaling pathway. Interestingly, HIV infection led to increased nuclear β-catenin in the HCT116 tumor cells (p<0.0001; Fig. 3d and e). If β-catenin signaling is activated, phosphorylated-β-catenin will be decreased and the nonphosphorylated active form of β-catenin levels will be increased, followed by subsequent β-catenin nuclear translocation. Western blot data confirmed that phosphorylated β-catenin at amino acid S33, S37, and T41 was decreased in the tumors from HIV-infected mice in comparison to noninfected mice (Fig. 3f ).
Maximally suppressed HIV replication results in decreased tumor growth
To further analyze how HIV contributes to tumor outgrowth, we extended our studies to determine the role of active versus antiretroviral therapy (ART)-suppressed HIV replication. Here, mice were treated with zidovudine, nelfinavir, and efavirenz (Fig. 4a). Such ART suppressed viral replication (p<0.05; Fig. 4b) and tumor size (p<0.05; Fig. 4c and d) and weight (p<0.05; Fig. 4e) in HIV-infected mice treated with ART in comparison to HIV-infected mice that were left untreated. Furthermore, IHC data confirmed that ART reversed the epithelial cell proliferation by BrdU staining (p<0.01; Fig. 5a and b) and β-catenin expression (p<0.05; Fig. 5a and c) observed in HIV-infected mice not treated with ART. Interestingly, these HIV-related tumor characteristics (Fig. 4b–e and Fig. 5a–c) were reversed with ART despite the fact that CD4+ T cell counts were not yet corrected by ART (Fig. 5d and e).
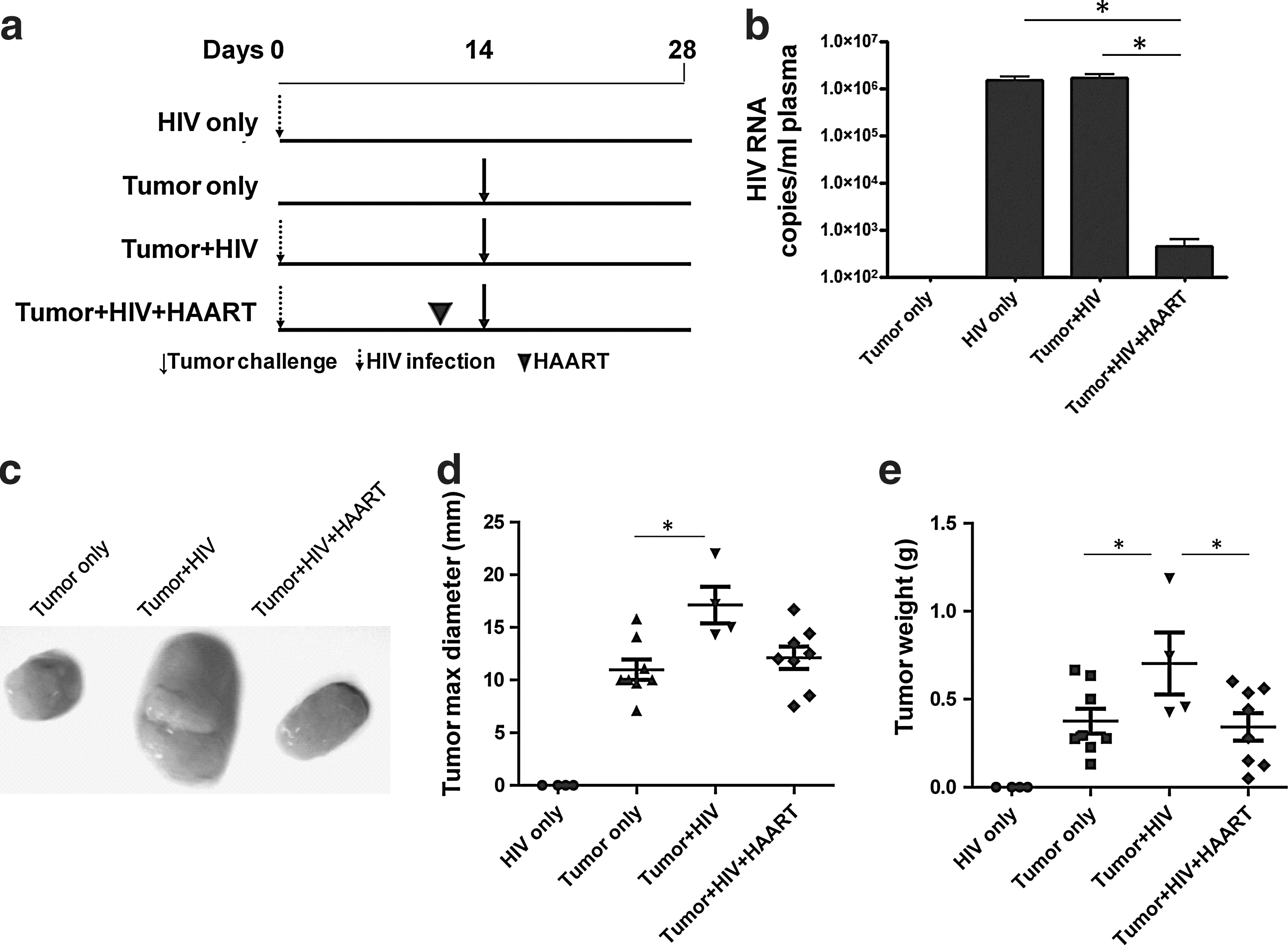
Reversal of tumor outgrowth in HIV-infected NSG-HuPBL mice after antiretroviral therapy.
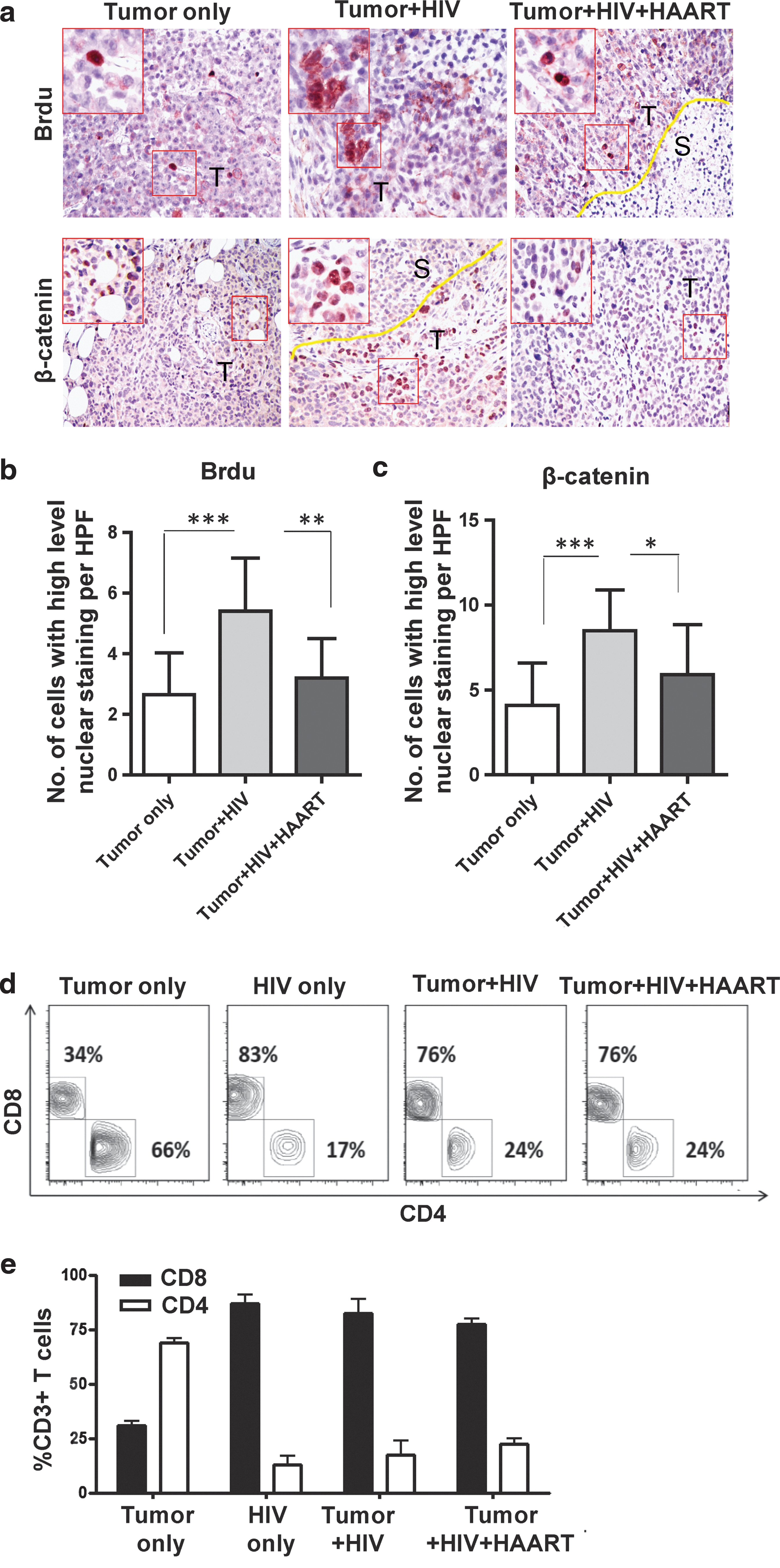
Reversal of tumor outgrowth pathological and β-catenin expression changes in HIV-infected NSG-HuPBL mice after antiretroviral therapy.
Discussion
There are significant clinical data indicating that HIV is a comorbid condition for tumorigenesis but the mechanism that drives this association is not entirely clear. Our findings indicate that uncontrolled HIV replication can accelerate colon cancer outgrowth. Specifically, we have demonstrated that HIV-infected humanized mice developed significantly larger and heavier colon cancer tumors than mice without HIV infection. In line with this enhanced tumorigenesis, epithelial cell proliferation was significantly enhanced in HIV-infected mice compared to mice without HIV infection and β-catenin activation was higher in HCT116 tumors in HIV-infected mice. Importantly, we found that ART-suppressed HIV replication resulted in decreased tumor outgrowth.
Several mechanisms may explain these observations, including (1) HIV induction of β-catenin within the tumor. β-Catenin plays a transcriptional role in modulating cell growth and proliferation in the intestine. 17 –20 Furthermore, β-catenin is associated with adenomatous polyposis coli (APC), a tumor suppressor, and oncogenic mutations of β-catenin genes have been found in most colon cancers with wild-type APC. 17,21 Such mutations initiate the neoplastic process, resulting in small benign tumors (adenomas), which progress as larger and more malignant tumors after accumulating mutations in other growth-controlling pathway genes (KRAS, BRAF, PIK3CA, or TP53). 17 (2) HIV-mediated inflammation that can create a microenvironment for accelerated tumor growth. In our study we used a transplantable tumor model to analyze tumor growth characteristics in the context of HIV infection. A related scenario in the clinical setting may involve individuals who have precancerous lesions or tumors that are dormant (by halted growth or equilibrium between tumor cell growth and immune response-driven cell death). Although our data for mouse macrophage marker CD68 in tumors did not show a significant difference in HIV-negative versus HIV-positive groups, HIV-mediated inflammation may directly (by supplying the tumor with growth factors) or indirectly (by decreasing the immune response) lead to the outgrowth of the tumor.
Intestinal epithelial cells are not infected by HIV but they respond to gp120 binding to CCR5 or CXCR4, both expressed on cells in the intestinal mucosa. 22–23 For a future study, we will investiagte whether gp120 is responsible for inducing epithelial tumor outgrowth through the β-catenin pathway.
While we acknowledge the limitations of our study, including the use of a transplantable human tumor cell line, subcutaneous tumor challenge, partial immune reconstitution afforded by adoptive transfer of adult PBLs, etc., we likewise advocate for the cutting edge approach that was utilized to investigate the progression of cancer in this humanized mouse model of HIV infection. The innovation of this model lies in its conceptual and technical framework of allowing the in vivo study of both HIV infection and cancer. From the perspective of HIV studies, the humanized mouse model has emerged as a significant tool with which to study responses to HIV and the mechanisms associated with HIV latency. 24,25 From the perspective of cancer studies, a recent report showed that melanoma metastasis in a humanized mice model correlates with metastasis probability in patients. 6 To our knowledge, our group is the first to adapt this model for a multidisciplinary approach assessing the impact of HIV on colon cancer progression and to define the molecular mechanisms by which it does so in vivo. We chose this xenograft cancer model for the current studies because (1) a humanized mice challenged with human colonic cancer cells allows for productive HIV infection and successful tumor challenge; (2) virus–host interactions in this model can be determined under well-controlled experimental conditions; (3) the readouts (e.g., tumor size and weight) are very straightforward; (4) the outcome can be achieved in an effective and efficient way (e.g., tumors can be analyzed 2 weeks post-HCT116 injection); and (5) humanization with PBLs (rather than fetal stem cells or thymus) allows us in future studies to utilize both PBLs and tumors derived from the same patients.
Overall, we hope our successes here spark further interest in the field for the use of this humanized model of HIV and cancer and expect it to provide a framework for understanding how HIV infection may inadvertently promote the development of cancer. Furthermore, we anticipate that our findings related to the role of β-catenin will make it possible to define the mechanisms by which HIV allows for the tumorigenesis of AIDS-defining cancers (ADCs) and non-AIDS-defining cancers (NADCs), and will afford the clinical field another lead in the treatment of colon cancer in the context of HIV infection. Lastly, our findings that antiretroviral therapy reversed tumor development in the context of HIV infection independent of CD4+ T cell return provide support for the concepts of early treatment of HIV patients even prior to CD4+ T cell decline and of increased cancer vigilance in HIV patients regardless of their CD4+ T cell recovery.
Footnotes
Acknowledgments
HIV-1Ba-L was obtained through the NIH AIDS Reagent Program, Division of AIDS, NIAID, NIH, which was contributed by Dr. Suzanne Gartner, Dr. Mikulas Popovic, and Dr. Robert Gallo.
Author Disclosure Statement
No competing financial interests exist.