
Abstract
Galectin-9 (Gal-9) is a β-galactosidase-binding lectin that promotes apoptosis, tissue inflammation, and T cell immune exhaustion, and alters HIV infection in part through engagement with the T cell immunoglobulin mucin domain-3 (Tim-3) receptor and protein disulfide isomerases (PDI). Gal-9 was initially thought to be an eosinophil attractant, but is now known to mediate multiple complex signaling events that affect T cells in both an immunosuppressive and inflammatory manner. To understand the kinetics of circulating Gal-9 levels during HIV infection we measured Gal-9 in plasma during HIV acquisition, in subjects with chronic HIV infection with differing virus control, and in uninfected individuals. During acute HIV infection, circulating Gal-9 was detected as early as 5 days after quantifiable HIV RNA and tracked plasma levels of interleukin (IL)-10, tumor necrosis factor (TNF)-α, and IL-1β. In chronic HIV infection, Gal-9 levels positively correlated with plasma HIV RNA levels (r=0.29; p=0.023), and remained significantly elevated during suppressive antiretroviral therapy (median: 225.3 pg/ml) and in elite controllers (263.3 pg/ml) compared to age-matched HIV-uninfected controls (54 pg/ml). Our findings identify Gal-9 as a novel component of the first wave of the cytokine storm in acute HIV infection that is sustained at elevated levels in virally suppressed subjects and suggest that Gal-9:Tim-3 crosstalk remains active in elite controllers and antiretroviral (ARV)-suppressed subjects, potentially contributing to ongoing inflammation and persistent T cell dysfunction.
Introduction
T
Inflammation has been linked as a potential mediator to this increase in non-AIDS-related mortality and morbidity among treated patients. 5,12 Multiple parameters of immune inflammation are elevated in untreated HIV infection, and decrease but fail to normalize during treatment. 13 –15 Significantly higher levels of interleukin (IL)-6, high-sensitivity C-reactive protein (CRP), D-dimer, sCD163, and type 1 interferon-α responses are seen in treated HIV patients compared to the uninfected general population. 15 –17 High levels of sCD14 (a marker of monocyte response to microbial lipopolysaccharide) are known to be associated with an increased risk of mortality as a result of microbial translocation into the periphery. 18,19 We and others have shown that CD8 T cell activation, as measured by the coexpression of HLA-DR and CD38, is higher in treated subjects compared to uninfected controls. 20 Strategies have focused on targeting several inflammatory pathways including interferon (IFN)-α and interleukin (IL)-7, however, they have not been successful in both human and primate studies. 21 –24 Recently antiinflammatory agents such as anti-IL-6, anti-tumor necrosis factor (TNF), choloroquine, methotrexate, and statins have either been tested or are advancing into clinical trials. 25 –29 Identification of novel mediators that participate in HIV-driven immune activation and inflammation that can be effectively targeted is essential.
In addition to elevated inflammation, CD8+ T cell dysfunction and immune exhaustion are common features of many chronic viral infections, including HIV and hepatitis C virus (HCV) infections. 30 –34 The mechanisms of T cell dysfunction are complex, but are in part mediated by a distinct set of inhibitory negative checkpoint receptors [T cell immunoglobulin mucin domain-3 (Tim-3), protein disulfide isomerases (PDI), and CD160]. 35 –37 T cell exhaustion has also been shown to be associated with HIV disease progression and suboptimal CD4+ T cell reconstitution in elite controllers and ARV-suppressed HIV-infected adults, respectively. 38,39
Galectin-9 (Gal-9) is a β-galactoside-binding lectin whose role has rapidly emerged, from what was once thought to be limited to being an eosinophil-specific chemotactic activity 40 to a complex role in either inhibiting or promoting various phases of the host immune responses. Gal-9 exhibits proinflammatory effects by promoting tissue inflammation, the maturation of monocyte-derived dendritic cells, and antimicrobial immunity. 41 –44 Gal-9 also functions as a negative immunomodulator by suppressing Th1 immune responses and the generation of IL-17-secreting CD4+ T cells (Th17) and by induction of regulatory T (Treg) cells. 45 –48 Gene and protein expression studies demonstrate that Gal-9 is present in a variety of cell types including endothelial cells, embryonic kidney cells, tumor cells, and Kupffer cells, 40, 49 –51 and among immune cells intracellular and surface forms of Gal-9 have been demonstrated on CD4+ T cells and mast cells. 52 –56 Gal-9 engages with numerous cell surface receptors such as Tim-3, a variety of PDIs, and IgE. 45,57,58 The consequences of Gal-9 interactions with these diverse receptors are now being unraveled.
The interaction of Gal-9 with the Tim-3 receptor on Th1 cells induces cell death and regulates CD8 T cell responses. 45,59 –61 We and others have observed that Tim-3+CD8+ T cells are increased in progressive HIV infection, and engagement presumably through a Gal-9 interaction leads to CD8+ T cell dysfunction. 33,60,62,63 During chronic HIV infection, Gal-9-expressing Treg cells are presumed to selectively suppress the proliferation of nonprotective HIV-specific CD8+ T cells with high Tim-3 levels, but not protective HIV-specific CD8+ T cells with low levels of Tim-3. 64 Soluble Gal-9 levels have been shown to be elevated in the plasma of HIV-infected subjects, 64 –66 suggesting that Gal-9 may play an important role in HIV pathogenesis. Engagement of Gal-9 with Tim-3+CD4+ T cells in vitro renders CD4+ T cells less susceptible to laboratory-adapted HIV isolates by downregulating HIV entry coreceptors, CCR5 and CXCR4, features that appear beneficial for viral clearance. 67 Recently, however, Gal-9 has been shown to facilitate HIV entry into CD4+ T cells through PDI in a Tim-3-independent manner and thus could result in increased viral replication. 58
Despite these findings, the dynamics of circulating Gal-9 levels during HIV infection and their roles in HIV pathogenesis remain poorly characterized. In this study, we investigated plasma Gal-9 levels during acute and chronic HIV infection and illustrate that elevations in circulating Gal-9 have the potential to play an important role in vivo during HIV disease pathogenesis and HIV-induced immune exhaustion.
Materials and Methods
Study subjects
Study blood specimens were as follows: (1) sequential cryopreserved plasma samples from 10 initially HIV-seronegative biweekly plasmapheresed donors who subsequently acquired HIV infection. 68 The sample time courses for each donor thus spanned time points from prior to plasma virus detection through seroconversion; (2) cryopreserved peripheral blood mononuclear cells (PBMCs) and plasma samples from selected participants in a San Francisco-based HIV-infected SCOPE cohort. The subjects studied included chronic HIV-infected noncontrollers (n=20), elite controllers (n=20) and ARV-suppressed subjects (n=20), and HIV-seronegative subjects (n=19) from SCOPE (Table 1); (3) plasma and PBMC samples from HIV-seronegative subjects obtained from age-matched subjects in the Hawaii Aging cohort. The study was approved by the institutional review boards of the University of California, San Francisco and the University of Hawaii. All individuals gave written informed consent and investigations were conducted according to the principles expressed in the Declaration of Helsinki.
IQR, interquartile range.
MTF, male to female.
HIV viral load (NC vs. EC, p<0.0001), (NC vs. AS, p<0.0001).
CD4+ T cell count (NC vs. EC, p<0.0001), (NC vs. AS, p=0.002).
CD4+ T cell nadir count (NC vs. EC, p=0.001), (NC vs. AS, p<0.0001), (EC vs. AS, p=0.0001).
CD8+ T cell activation (NC vs. EC, p<0.001), (NC vs. AS, p<0.0001).
Viral load assessment
HIV viral loads were retrospectively analyzed by Quest Diagnostics by using a Roche Amplicor HIV Ultra assay with a lower limit of detection of 50 RNA copies/ml (Roche Diagnostic Systems, Branchburg, NJ).
Gal-9 ELISA assay
In brief, 96-well plates (Nunc, Naperville, IL) were coated with an antihuman Gal-9 monoclonal antibody (mAb) (9S2-3, GalPharma, Kagawa, Japan), blocked with 3% fetal bovine serum containing 0.05% Tween 20 in phosphate-buffered saline (PBS), then incubated for 1 h at 37°C with 8-fold-diluted plasma. After several washings, Gal-9 remaining in the wells was recognized by polyclonal antihuman Gal-9 antibody conjugated with biotin using EZ-Link Sulfo-NHS-Biotin reagent (Pierce, Rockford, IL). Quantification was performed using streptavidin-conjugated horseradish peroxidase (Invitrogen, Tokyo, Japan) and the colorimetric substrate tetramethylbenzidine (KPL, Gaithersburg, MD), and the optical density was read with a microplate spectrophotometer (Bio-Rad, Hercules, CA).
Peripheral blood mononuclear cell staining and flow cytometric analysis
PBMCs were washed with PBS supplemented with 1% bovine serum albumin (BSA) and 2 mM EDTA before staining. Cells were stained with the following mAbs: ECD-conjugated anti-CD3 (Beckman Coulter, Brea, CA), Qdot605-conjugated anti-CD8 (Invitrogen, Carlsbad, CA), Alexa 700-conjugated or FITC-conjugated anti-CD4, and Alexa 700-conjugated anti-Tim-3 (R&D Systems, Minneapolis, MN). An amine aqua reactive dye (AARD) (Invitrogen) was used to exclude dead cells. Fluorescence minus one (FMO) samples were prepared for detecting any spillover from other channels. An isotype IgG control was used to facilitate gating. All cells were fixed with 1% paraformaldehyde (PFA) and collected using a 4-laser custom BD For tessa flow cytometer (BD Biosciences, San Jose, CA). A total of 100,000 cells were collected and analyzed with FlowJo software (TreeStar, Ashland, OR).
Surface plasmon resonance spectroscopy
The interaction between Gal-9 and Tim-3 was tested by using both an antihuman Fc capture surface and a Gal-9 surface using a Biacore T200 biosensor (GE Healthcare).
Gal-9 surface
As it was theorized that the interactants being studied here may interact with the dextran present on the CM-series sensor chips, which contain carboxymethylated dextran, a Gal-9 surface was prepared on a C1 chip. The Gal-9 was coupled through primary amines by using the amine coupling procedure. To investigate binding of Tim-3 to Gal-9, a solution of 1 μg/ml (21 nM) Tim-3 human IgG Fc chimera (R&D systems, 2365TM) was injected over the Gal-9 surface. The binding level of this initial injection was about 8 response units (RU). A theoretical binding level of up to 15–32 RUs was anticipated assuming a 1:1 binding. A second injection of 5 μg/ml Tim-3 was performed, resulting in a final binding level of about 15 RU. No nonspecific binding was observed on the reference surface.
Tim-3 surface
The antihuman Fc capture surface was prepared on a CM5 sensor chip, which is composed of carboxy-methylated dextran. As it was possible that the interactants being studied here would interact with the dextran present on the CM-series sensor chips, an initial test was performed to access the nonspecific binding to the CM5 chip. Solutions of 10 μg/mL Gal-9 and Tim-3 were injected over a CM5 chip containing an unmodified CM5 sensor surface. A CM5 chip was coupled with 1.82 ng of Tim-3 human IgG Fc chimera at pH 4.5 (equivalent to 1,820 RU). Recombinant human Gal-9 (R&D Systems 2045GA) was injected over the Tim-3 surface in 2-fold serial dilutions from 563 nM to 18 nM, and the change in response units was measured. A 7 min injection of 20 μg/ml solution of Gal-9 in 10 mM sodium acetate pH 5.5 resulted in an immobilization level of 12–25 RU. Initial assessment indicated that there was little to no nonspecific binding. A multicycle kinetic assay was performed to provide information regarding both the kinetics and affinity of the interaction between Tim-3 and Gal-9.
Statistical analyses
Statistical analyses were performed by using SAS System 9 for Windows XP (SAS Institute, Cary, NC) and Prism (GraphPad Software, San Diego, CA). Nonparametric statistical tests were used. The Mann-Whitney U test was used for comparison tests and the Spearman rank test was used for correlation analyses. For paired samples, a paired t-test was used to compare different groups.
Results
Circulating levels of Gal-9 are rapidly elevated in acute HIV infection
Sequential samples collected from plasma donors acquiring HIV infection present a rare opportunity to evaluate the dynamics of circulating Gal-9 levels during the earliest stages of HIV infection. We measured the magnitude and duration of alterations in Gal-9 levels in the plasma of 10 subjects before (two time points) and after (five time points spanning 14–42 days) detectable viremia using a Gal-9 ELISA. 51 Plasma HIV RNA levels increased rapidly in all subjects (Fig. 1A), with the exception of subject 9032 whose viral load peaked at just over 3 log10 copies RNA/ml and appeared to subsequently decline. Gal-9 levels rose during this period in all subjects (Fig. 1B), although Gal-9 levels in subject 9021 (gold line) rose very little, despite robust viral replication.

Kinetics and levels of Gal-9 in early HIV infection. The graphs display
We next evaluated the timing relative to T0 (the time point when viremia first reached 100 RNA copies/ml) of the first elevation in the plasma level of Gal-9 levels and assessed the temporal relationship with 18 cytokines previously quantitated in this cohort. 68 The median time for the first elevation of Gal-9 in the 10 subjects studied was day 5 (median peak of 57.4 pg/ml) (Fig. 1B), similar to the timing with which the first elevations in plasma cytokines including IP-10, IL-15, TNF-α, IFN-α, MCP-1, and IL-1R in this cohort, collectively referenced as the first wave of the cytokine storm, occurred. 68 Gal-9 levels closely tracked several cytokines including IL-1β, IL-10, and TNF-α before and after quantifiable HIV viral load (Fig. 2A, C, and E). Taken together, these data benchmark Gal-9 with the earliest soluble factors released in response to HIV infection.
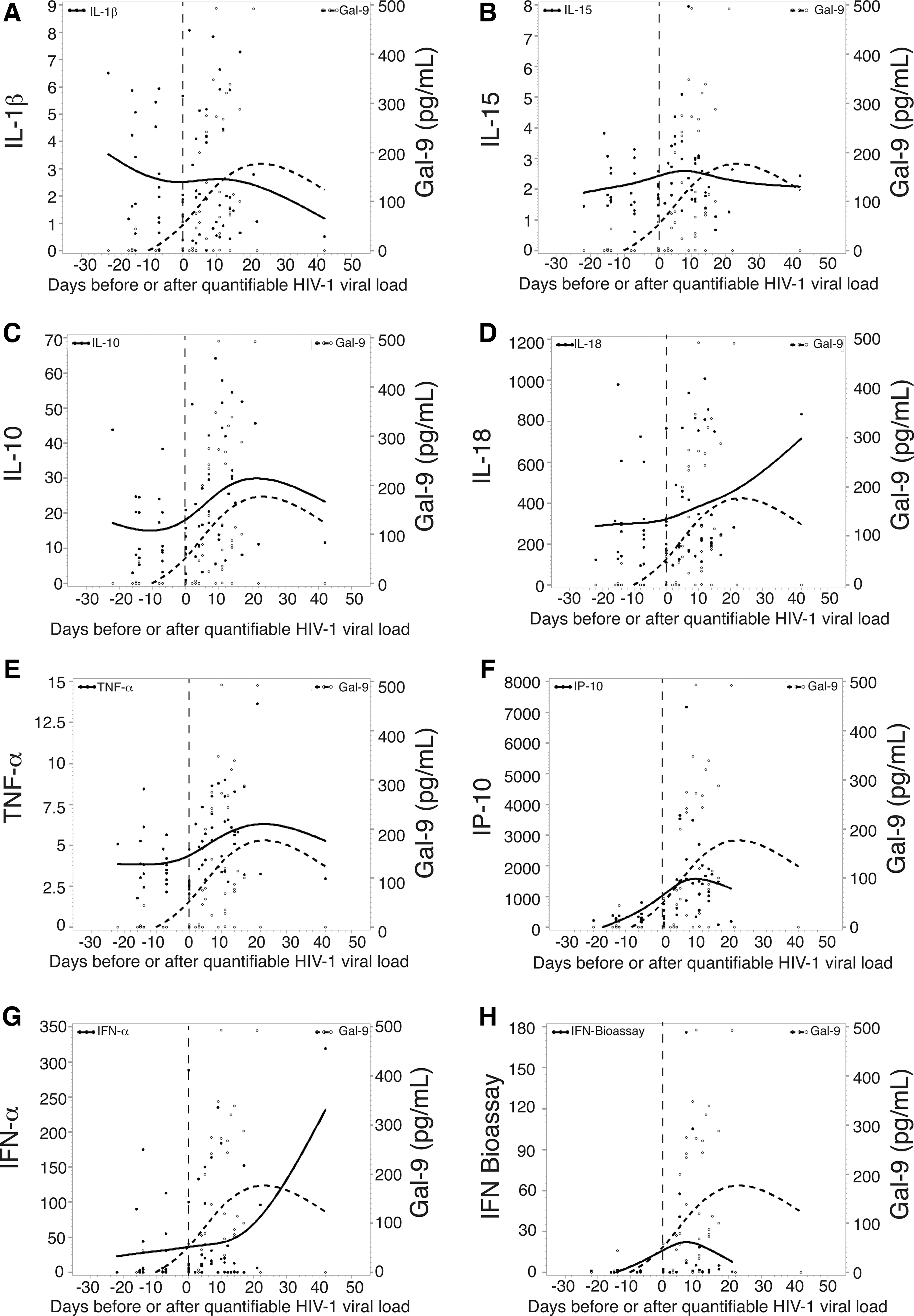
Mean change relative to baseline in plasma analyte levels over time in a group of subjects infected with HIV.
Elevated level of Gal-9 in chronic HIV infection despite suppressive ARV therapy or elite HIV control
To assess the effects of HIV on Gal-9 levels in chronic infection we evaluated the levels of circulating Gal-9 in 20 HIV controllers in the absence of treatment (“elite” controllers), 20 untreated subjects with high viremia (noncontrollers), 20 long-term treated subjects with undetectable HIV levels (“ARV-suppressed”), and 19 matched uninfected controls. As a group, plasma Gal-9 levels in chronic HIV-infected subjects [median, 325.6 pg/ml; interquartile range (IQR) (193.6, 655.6)] were significantly elevated compared to controls (54 pg/ml; 16, 181) (Fig. 3A). When the HIV-infected subjects were segregated we observed that noncontrollers had the highest Gal-9 levels (556.6 pg/ml; 288.3, 877.2) and were significantly higher compared to healthy subjects (Fig. 3B). Elite controllers and ARV therapy-suppressed subjects had significant higher levels of Gal-9 than HIV-seronegative controls (Fig. 3B). Despite viral control, elite controllers and ARV therapy-suppressed subjects had significantly greater levels of Gal-9 compared to HIV-seronegative controls, suggesting that high levels of Gal-9 are maintained in the circulation and do not normalize despite viral control.
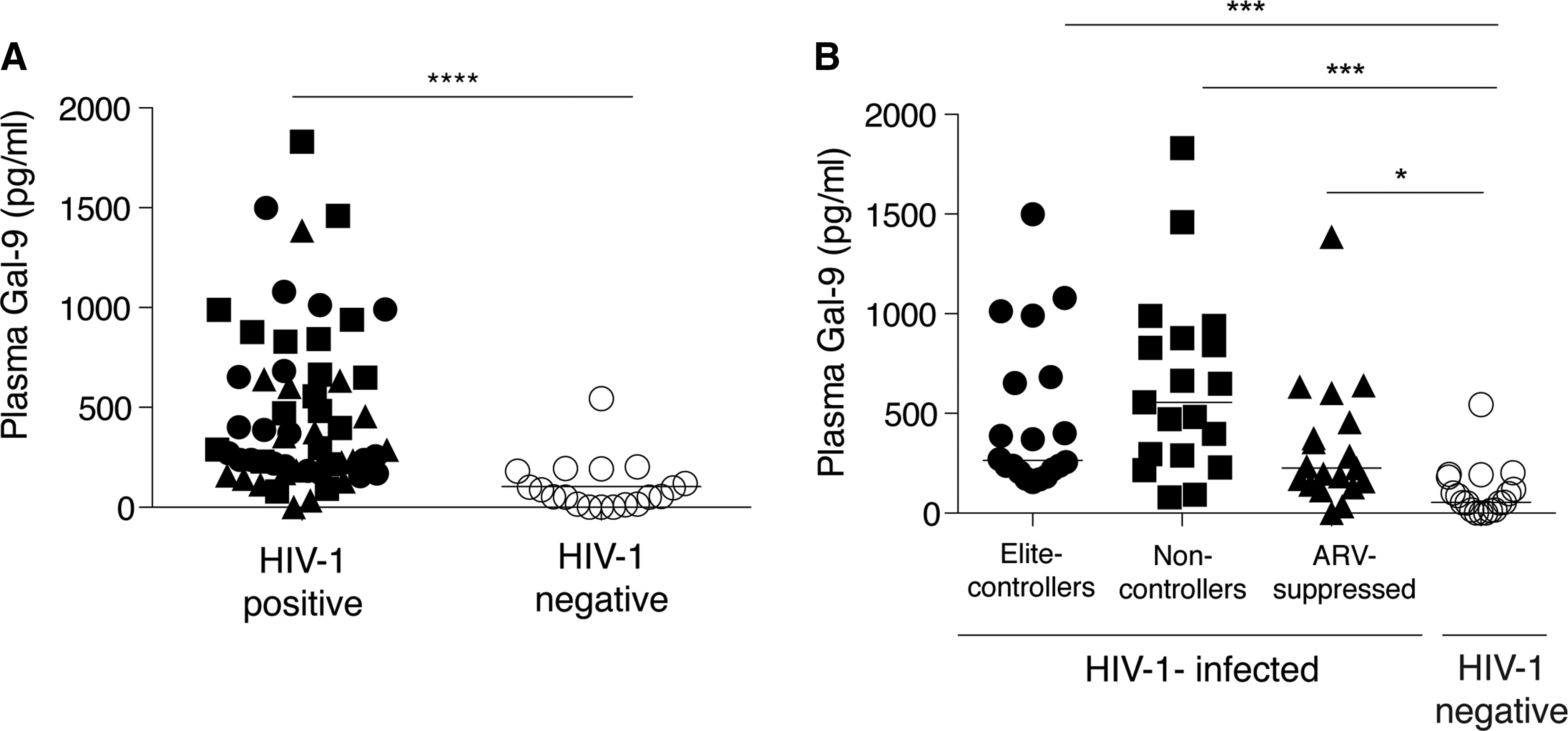
Gal-9 levels in chronic HIV infection.
Dynamics of expression of the Gal- 9 receptor, Tim-3, on CD8 T cells from HIV-infected subjects
Tim-3 has been described as a receptor for Gal-9,
45,62
although this was challenged in a recent study.
69
By using surface plasmon resonance (SPR), binding tests for Tim-3 and Gal-9 interaction were performed. A 10 μg/ml solution of Tim-3 human IgG Fc chimera was injected over an antihuman Fc surface, resulting in capture of about 720 RU of Tim-3. This was immediately followed by injection of 10 μg/ml (280 nM) Gal-9 and binding of about 1,890 RU was observed. A multicycle kinetic assay was performed to provide information regarding both the kinetics and affinity of the interaction. Using a 2-fold concentration series of Gal-9 from 280 nM to 1.1 nM, and including several blanks (0 nM) injected over the surface containing Tim-3 in duplicate, confirmed the Gal-9:Tim-3 interaction (Supplementary Fig. S1; Supplementary Data are available online at
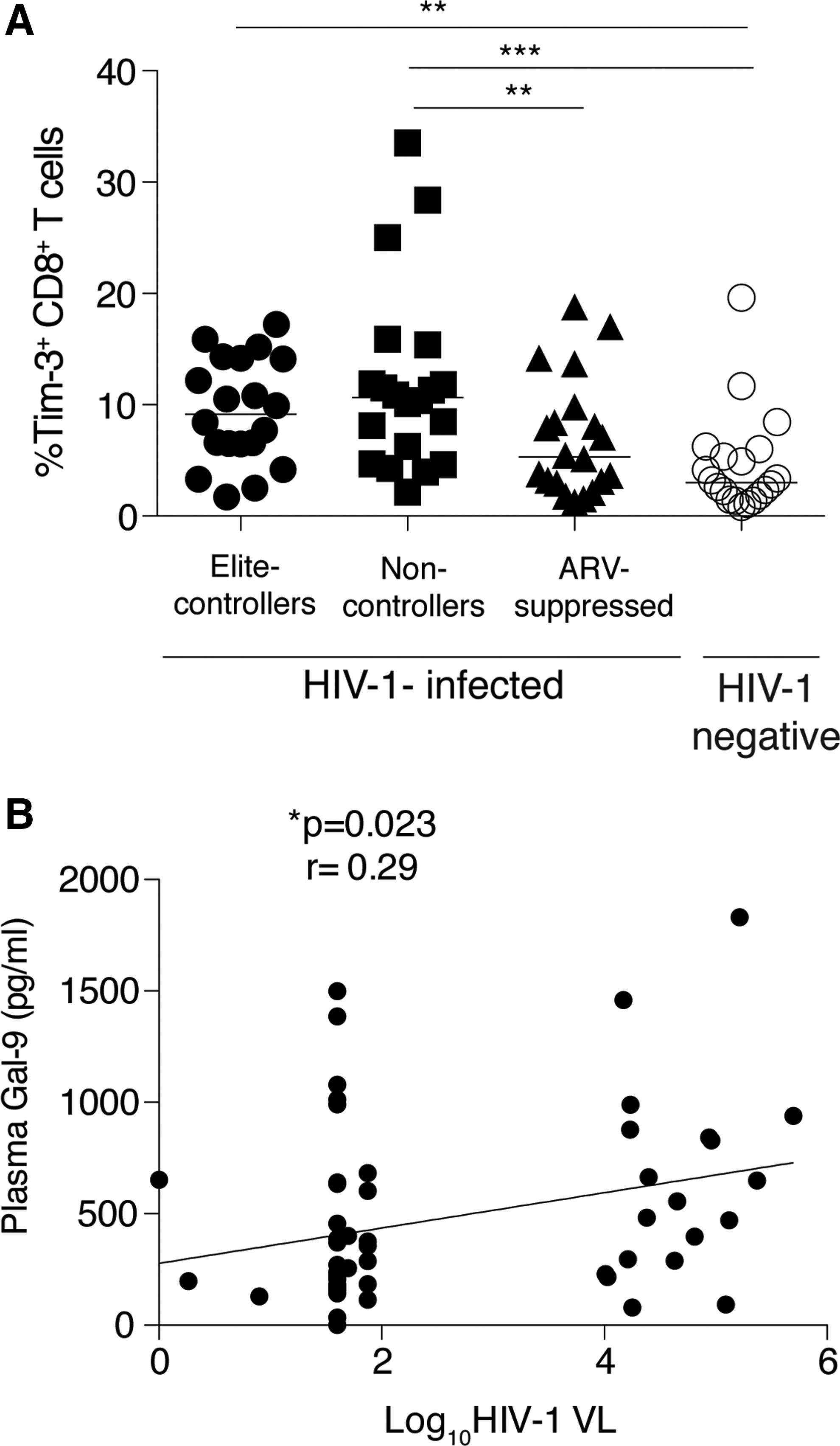
Tim-3 expression on T cells of HIV-infected and HIV-uninfected subjects and correlation between Gal-9 levels and HIV viremia.
Association of circulating plasma Gal-9 and HIV plasma viremia
We have previously observed that Tim-3+CD8+ T cells positively correlated with plasma viral load. 63,70 We performed a linear regression analysis in this cohort and observed a positive correlation between the frequency of Tim-3+CD8+ T cells and HIV plasma viral load in the overall cohort (p=0.03, r=0.26) (Supplementary Fig. S2). Furthermore, we observed a positive correlation between Gal-9 levels and plasma HIV viral load (p=0.023, r=0.29) in the overall cohort (Fig. 4B) but not when segregated into nonviremic or viremic groups. No associations were observed with Gal-9 levels or Tim-3+CD8+ T and CD4 count or between Tim-3+CD8+ T and Gal-9 levels (data not shown). Overall, these results suggest that Gal-9 release may be driven in part by viral replication.
Discussion
We performed a comprehensive study of the immunomodulatory molecule Gal-9 in HIV infection. We found that in acute HIV infection, Gal-9 increased rapidly after the first detection of viremia with the earliest median time for the first elevation being 5 days postviremia, comparable to the timing of the elevation in the first wave of other soluble factors previously evaluated at postviremic time points in this cohort. 68 Gal-9 tracked with viremia and cytokines (IL-1β, IL-10, and TNF-α) in acute HIV infection and remained elevated during chronic infection, even in subjects exhibiting elite viral control. Compared to HIV-negative controls, we observed an elevated frequency of Tim-3 expressing CD8+ T cells in HIV-infected subject with undetectable HIV viral load (both ARV-suppressed subjects and elite controllers). In addition, we show that there is a positive correlation between plasma Gal-9 levels and HIV viral load, suggesting that this interaction may be in part driven by HIV viral replication. Collectively, these data suggest that Gal-9 may drive HIV pathogenesis by rendering CD8 T cells dysfunctional as well as contribute to ongoing persistent inflammation in the setting of viral suppression.
Acute HIV infection is characterized by virus expansion at the transmission site and in local lymphoid tissues followed by systemic dissemination and an exponential increase in viremia. 71 The rise in plasma HIV RNA is associated with a massive depletion of memory CD4+ T cells, mainly from gut-associated lymphoid tissues (GALT). 72 During this acute phase, elevated levels of cytokines such as IFN-α, IL-15, IP-10, TNF-α, MCP-1, IL-6, IL-18, and IFN-γ give rise to a “cytokine storm” that is unique to HIV, 68 and may also contribute to CD4+ T cell apoptosis. 73 Gal-9 can be induced by IL-1β, IFN-γ, and double-stranded RNA. 74 Overall, our data as a group indicated that Gal-9 is a plasma-soluble factor that may serve as one of the earliest markers after viral emergence with a potential role in CD4+ T cell loss and HIV viral entry. 58 The dynamics of Gal-9 levels were variable and not absolute during acute HIV viral evolution and this may be attributable to differences in host or viral factors in some individuals as has been observed with the evolution of other cytokines such as IFN-α. 68 However, it will be interesting to determine whether Gal-9 also has the potential to act as one of the earliest markers in other viral infection such as dengue virus, hepatitis C virus (HCV), or hepatitis B virus (HBV), or whether this feature is unique only to HIV infection and HIV variants.
Furthermore, given the immunosuppressive functions of Gal-9 45,46,48 and the increased frequency of Tim-3+CD8+ T cells, we would propose the function of Gal-9 in T cell-driven immune exhaustion is likely occurring during acute infection. We and others have previously shown that HIV infection leads to the preferential expansion of CD4+ Treg cells and loss of Th17. Furthermore, in vitro studies have found that Gal-9 induces differentiation of naive T cells to Treg cells, and suppresses differentiation to Th17 cells. 47,48,75 Given the multiple functions of Gal-9 in regulating CD4+ T cell responses, 48 this may be in part driven by Gal-9. 48,76 –78 Overall, our results demonstrate that Gal-9 levels are an early biomarker of HIV infection and likely track HIV viremia through primary infection and then persist into chronic infection even in subjects exhibiting good viral control.
HIV-infected subjects on suppressive ARV therapy exhibit elevated levels of circulating inflammatory cytokines in plasma that appear to be linked to non-AIDS illness. 5 The elevated levels of Gal-9 in chronic HIV infection despite viral suppression suggest that Gal-9 may contribute to ongoing immune inflammation. We observed that despite viral suppression, Gal-9 levels remained elevated compared to both uninfected demographically and age-matched controls. Despite undetectable viremia, unspliced HIV RNA appears to serve as a predictive marker for the outcome of ARV therapy. 79 The elevated Gal-9 level in ARV-suppressed subjects may be attributed to the presence of unspliced cellular HIV RNA. Soluble Gal-9 levels have been shown to be elevated in the plasma of HIV-infected subjects, 65 –67 and Jost et al. 66 have shown that despite ARV therapy, chronic HIV-infected adults did not experience declines in plasma Gal-9 levels when compared to untreated chronic HIV-infected subjects.
Elite controllers maintaining a viral load of <50 copies/ml represent less than 1% of all untreated HIV-infected patients. 80 HIV-specific cytotoxic T lymphocytes (CTL) are preferentially primed for apoptosis and this represents a viral escape mechanism 81 ; however, HIV-specific CD8+ T cells from elite controllers are primed for survival. 82 The majority of elite controllers have very low rates of CD4+ T cell decline and progression of disease. 83 However, low CD4+T cells have been observed in a few elite controllers. 84 Some elite controllers with high levels of immune activation immunologically progress to AIDS despite maintaining virologic control without ARV therapy. 11,84 Moreover, elite control can be a temporary state with some patients experiencing loss of virologic control and/or CD4+ T cell decline over the course of infection. 10,11
Few studies have evaluated soluble mediators in elite controllers that could in part explain the heterogeneity of this group of patients. We and others have found an unexpectedly high degree of coronary atherosclerosis and elevated markers of immune activation in elite controllers compared to HIV-negative controls. 85 Studies from our group and Pereyra et al. suggest that elite controllers have low levels of viremia suggesting a basal level of HIV replication. 86,87 From our current results, we show that elite controllers have elevated levels of Gal-9 and significantly higher Tim-3+CD8+ T cells compared to uninfected controls, suggesting that Gal-9:Tim-3 crosstalk persists in this virally suppressed group leading to impaired T cell function and CD4+ T cell loss. A higher level of Gal-9 in the midst of viral suppression is indeed intriguing. In line with our findings, others have shown that ARV therapy did not decrease the levels of Gal-9 in a chronic HIV-infected adult study group as a whole when compared to untreated chronic HIV-infected subjects. 66 We and others have recently shown that a low level of HIV viral replication occurs in elite controllers and instituting ARV therapy in this population leads to a significant decline in residual HIV viral replication and levels of immune activation, arguing for instituting ARV therapy in this population, previously regarded as not requiring ARV treatment. 86,87 These data postulate that low level replication is ongoing in these subjects.
Our results indicate that there is a difference in Tim-3 expression on CD8+ T cells between elite controllers and healthy individuals, but not those on antiretroviral therapy, which may potentially be because of the fact that elite controllers show elevated markers of immune activation and bacterial translocation compared to ARV-suppressed patients. 88,89 Moreover, we and others have also shown that elite controllers have a higher median plasma viral load than that usually documented in ARV-suppressed patients. 86,87,90
HIV infection results in the depletion of CD4+ T cells. 72 The precise mechanism by which HIV depletes CD4+ T cells is complex and unresolved. Recently, indirect cell killing involving abortive HIV infection of CD4+ T cells has been shown to be a potential cause of the vast majority of cell deaths occurring in lymphoid tissue. 91,92 Moreover, host-derived factors such as TNF-α, Fas ligand, and TRAIL, and viral factors such as HIV Tat, Vpr, and Nef released from infected cells have also been proposed for CD4+ T cell killing. CD4+ T cells are susceptible to HIV infection, and two recent in vitro studies have highlighted opposing roles for Gal-9 in HIV infection; one study revealed the novel property of viral entry in CD4+ T cells via Gal-9-induced retention of PDI on CD4+ T cells. 58 Another study has shown that Tim-3-expressing CD4+ T cells upon ligation with Gal-9 makes the cells less susceptible to HIV by decreasing the expression of CCR5 and CXCR4. 67 Furthermore, in vivo studies will be needed to clarify the role of Gal-9 in regulating HIV viral entry and susceptibility.
In summary, we show here that Gal-9 is markedly elevated soon after detectable viremia and is one of the earliest first wave factors in the cytokine storm of acute HIV infection. Gal-9 levels remain elevated despite viral suppression in chronic infection. We propose a model in which high levels of Gal-9 contributing to CD8+ T cell dysfunction through Tim-3 interactions are active during viral suppression even among elite controllers and may contribute to persistent inflammation. Targeting Gal-9 either by manipulating the secretory pathways or directly by competitive blockade may represent novel therapeutic approaches to suppress HIV-driven T cell immune exhaustion and inflammation.
Footnotes
Acknowledgments
We are deeply appreciative to the subjects of the SCOPE cohort for their participation in this study. This work was supported in part by the Creative and Novel Ideas in HIV Research Program (CNIHR) through a supplement to the University of California San Francisco-Gladstone Institute of Virology & Immunology Center for AIDS Research (UCSF-GIVI CFAR) funding (P30 A1027763). This funding was made possible by collaborative efforts of the Office of AIDS Research, the National Institutes of Allergies and Infectious Diseases (NIAID), and the International AIDS Society, Grant 2G12RR003061-26 from the National Center for Research Resources and grant 8G12MD7601-27 from the National Institute on Minority Health and Health Disparities. Research reported in this publication was also supported by the NIAID of the NIH and by the Center for HIV/AIDS Vaccine Immunology (CHAVI), grant U19-AI067854-07. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. The SCOPE cohort was supported in part by the NIAID (RO1 AI087145, K24AI069994), the UCSF Clinical and Translational Research Institute Clinical Research Center (UL1 RR024131), and the CFAR Network of Integrated Systems (R24 AI067039). We thank Cynthia Shuman, PhD (GE Healthcare) for Biacore application assistance.
Author Disclosure Statement
No competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.