
Abstract
CRF01_AE and subtype B are the two of major HIV-1 clades circulating in China. HIV spread more rapidly among men who have sex with men (MSM) than among populations with other risk behaviors. In Jiangsu province in China, the HIV-1 incidence among MSM was more than 3.8%. Our previous study showed that almost equal proportions of CRF01_AE, B, and CRF07_BC were circulating among MSM. Moreover, many kinds of CRF01Bs have been identified among MSM in Southeast Asia in recent years. It is therefore inevitable that recombination between CRF01_AE and subtype B will emerge among MSM in Jiangsu province in China. Here we identify a novel recombinant of CRF01_AE/B that has a distinctly different genome structure from other CRF01Bs and unique recombinant forms (URFs) previously identified. An analysis of the near full-length sequence of JS2010001 showed that it is composed of at least three interlaced CRF01_AE and B segments. Recently, many kinds of URFs and C circulating recombinant forms (CRFs) have emerged among MSM in China within a short period of time, which suggests that dual infection of HIV-1 among MSM in China is very common and that more effective intervening measures to prevent the spread of HIV among MSM should be taken.
T
CRF01_AE originated in central Africa in the 1970s and spread in Thailand in the 1980s through heterosexual transmission. 6 It was subsequently confirmed as the first large-scale epidemic of a recombinant HIV-1 strain in the world. In the early 1990s, CRF01_AE strains were first identified in China among people at risk for sexual transmission and injecting drug users (IDUs) in Yunnan Province and Guangxi Zhuang Autonomous Region, which is located in the southwest of China and borders Myanmar and Vietnam. 7,8 Subsequently, CRF01_AE strains spread quickly in China, accounting for approximately 28% of nationwide HIV infections in 2006, and becoming the predominant subtype. 9 More recently, CRF01_AE strains have also become the predominant strain among men who have sex with men (MSM) in China.
Subtype B′, the Thailand variant of subtype B, is also referred to as Thai B. 10 It was originally identified among IDUs in Thailand, and spread into neighboring regions including Myanmar, Malaysia, eastern India, and China. 11 It plays a unique role in the genesis of the HIV-1 epidemic in China. Subtype B′ is a single founding strain responsible for HIV-1 outbreaks among former plasma donors (FPD) in central China. In 2005, about 70,000 people living with HIV had been infected through unhygienic plasma collection from the early to the mid-1990s. 12 Subsequently, HIV subtype B began to spread through heterosexual or homosexual transmission.
Among the newly diagnosed HIV-positive population, MSM has been one of the major populations in China since 2009. Sentinel surveillance data also clearly demonstrated the rapidly increasing prevalence of HIV-1 among MSM. Dual infection in one person with HIV-1 among MSM is common due the lack of protective sex behavior and multiple sexual partners. 13 Therefore, the HIV-1 recombinant is more likely among MSM than among other at-risk populations in China.
Jiangsu province is located in eastern China. The first HIV case was recorded in 1992. By the end of September 2012, 8,960 HIV-1-positive cases had been reported. Among the HIV cases reported in 2012, 93.6% of them acquired their HIV infection through sexual transmission. Of sexual transmissions, homosexual transmission accounted for 48%. Now the high incidence of HIV occurred among MSM in Jiangsu province. 14 –16 Our previous study showed that CRF01_AE and the B subtypes were the two major HIV forms circulating among MSM in Jiangsu province. 16 Recently, many recombinants between CRF01_AE and B have been reported in many regions of the world. 3,4,17 –20 To determine whether the recombination events appeared among MSM in Jiangsu province, we enrolled HIV-positive MSM who were diagnosed in 2010 to conduct this study.
After obtaining written consent information, we spoke with the MSM in a private room and completed a structured questionnaire. Then 5 ml of whole blood was drawn using an EDTA-K3 vacuum tube (BD Company, USA). The plasma sample was separated by centrifugation at 3,000 rpm for 15 min. HIV antibody was screened using an ELISA kit (BioMerieux Ltd., France) and the positive samples were confirmed using a Western blot assay (Genelabs Diagnostics Pte Ltd., Singapore). Viral RNA was extracted from 140 μl of plasma with a QIAamp Viral RNA Mini Kit (Qiagen, GmbH, Germany) and reverse transcribed into cDNA using a SuperScript First-Strand Synthesis System for reverse transcription polymerase chain reaction (RT-PCR) (Invitrogen, USA) according to the manufacturer's instructions. Reverse transcription of the RNA was performed by priming with UNINEF7′ (GCACTCAAGGCAAGCTTTATTGAGGCTT, HBX2: nt 9,605–9,632). The amplification of HIV fragments was performed based on the previous method. 20 The following primers were used in the first round PCR: UNINEF7′, pro5F (AGAAATTGCAGGGCCCCT AGGAA, HBX2: 1,996–2,018), msf12b (AAATCTCTA GCAGTGGCGCCCGAACAG, HBX2: 623–649), and RT3474R (GAATCTCTCTGTTTT CTGCCAGTTC, HBX2: 3,453–3,477) to amplify HIV fragments. The primers were used in the second round PCR as follow: ENVinF1 (TGGAAGCATCCRGGAAGTCA GCCT, HBX2: 5,861–5,884), nefyn05 (GTGTGTAGTTCTGCCAATCAGGGAA, HBX2: 9,157–9,181), OLinF1 (AGGACCTA CRCCTGTCA ACATAATTGG, HBX2: 2,483–2,509), VIF-VPUinR (CTCT CATTGCCACTGTCTTCTGCTC, HBX2: 6,207–6,231), f2nst (GCGGAGGCTAGAAGGAGAGAGATGG, HBX2: 769–793), and proRT (TTTCCCCACTAACTTCTGTATG TCATTGAC A, HBX2: 3,308–3,338).
PCR products were purified using a gel extraction kit (Qiagen, GmbH, Germany) and then sequenced on an ABI 3730X DNA analyzer using BigDye terminators (Applied Biosystems, Foster City, CA). The sequences were blasted with all the sequences obtained in the laboratory for confirmation with previous results and to avoid possible cross-contamination. The sequences determined in this study are available in GenBank under accession number KF758551.
DNA sequences were assembled using Contig software. The sequences were blasted with all the sequences obtained in the laboratory for confirmation with previous results and to avoid possible cross-contamination. A codon-aligned nucleotide sequence alignment was constructed using the Gene Cutter tool from the Los Alamos HIV sequence database (
Nucleotide sequences were aligned manually using MEGA version 5.1, with HIV-1 reference subtypes and CRFs from the HIV database (
Using phylogenetic analysis, the near full-length sequences of JS2010001 form a monophyletic branch supported by a bootstrap value of 100%, separate from other global subtypes and known CRFs in the world (Fig. 1). SimPlot analysis reveals that the breakpoint corresponded to HXB2 nucleotide positions 2,759 and 4,876 (Fig. 2). Phylogenetic analysis of the subgenomic fragments confirms the two breakpoints identified (Fig. 3). The genomic fragments 769 to 2,758, 2,759 to 4,875, and 4,876 to 9,181 branched with references CRF01_AE, B, and CRF01_AE, respectively. The breakpoints are completely different from the CRF01B previously reported.
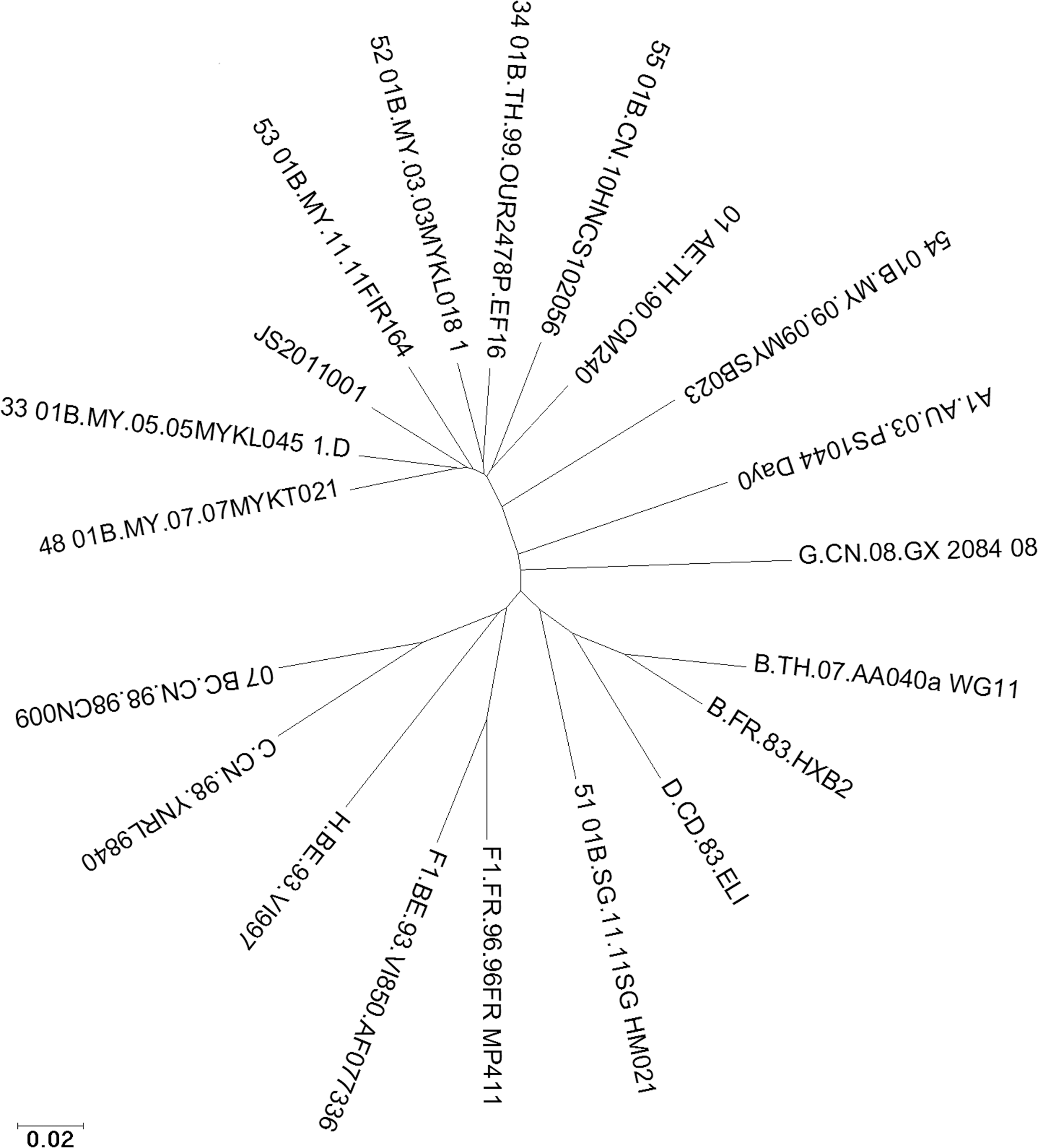
The phylogenetic tree of near full-length sequences showing the relationship between JS2010001 and other circulating recombinant forms and subtypes of HIV-1 previously identified.
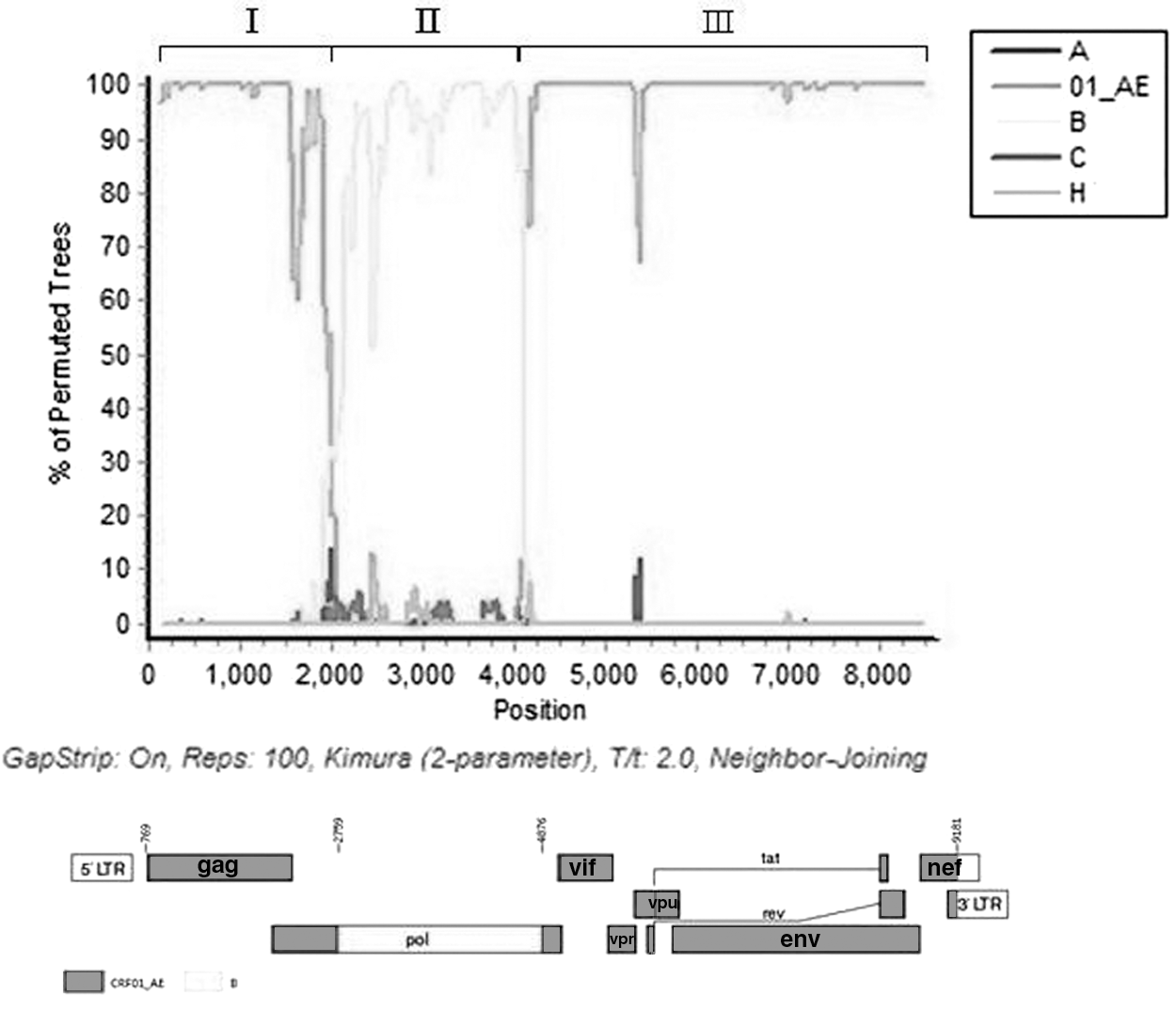
Bootscanning analysis of the near full-length sequence of JS2010001. Top: bootscanning plot of JS2010001 using A1 (PS1044_Day0), B′ (AA040a_WG11), CRF01_AECM240), C (YNRL9840), and H (VI997) as references. Bottom: mosaic map of JS2010001. Breakpoint positions were numbered according to the HXB2 numbering engine (

Phylogenetic analyses of three mosaic segments defined by bootscanning. The phylogenetic trees of three mosaic segments were constructed with MEGA 5.1 using the neighbor-joining method. The stability of the nodes was assessed by bootstrap analysis with 1,000 replications, and only bootstrap values of >75 were shown at the corresponding nodes.
In genomic regions I and III, JS2010001 forms a cluster distinct from the reference CRF01_AE, supported by a strong bootscan value of 100%. In genomic region II, JS2010001 forms a cluster distinct from the reference subtype B, supported by a strong bootscan value of 96%.
The recombinants of CRF01_AE and the B subtype have been reported in some regions of China, 3 –5,19 and they were all formed in the MSM population. Currently, sexual transmission is the predominant mode of HIV-1 transmission in most regions of China. Among the 7.8 million people living with HIV-1 in 2011, 46.5% were infected by heterosexual transmission and 17.4% were infected by homosexual transmission. The incidence of HIV-1 increased among MSM more rapidly than among populations with other risk behaviors. The proportion of cases resulting from sexual transmission increased from 33.1% in 2006 to 76.3% in 2011. The proportion arising from homosexual transmission increased from 2.5% in 2006 to 13.7% in 2011. Of MSM 85% have engaged in sexual activity with multiple homosexual partners within the past 6 months, and only 43% of MSM consistently use condoms during anal sex. Even more, 31.8% of them had regular female partners and 25.9% were currently married to a woman. MSM have played a bridge role in spreading HIV-1 among various populations. Therefore, the multiplicity of infection in HIV is more likely to occur among MSM, which leads to the formation of the new recombinant between subtype and CRF.
In summary, we have identified a novel recombinant form derived from CRF01_AE and subtype B among MSM. Phylogenetic analysis showed that it has the distinct genomic structure of other CRF_01B and URFs reported. Many types of URFs and CRFs have emerged among MSM in a short period in China, which suggests that more effective intervention measures to prevent the spread of HIV-1 among MSM should be taken, and highlights the importance of ongoing molecular epidemiological surveillance among MSM in China.
Sequence Data
The near full-length genomic sequence of JS2010001 has been submitted to GenBank with accession number KF758551.
Footnotes
Acknowledgments
This work is supported by Jiangsu Province's Key Provincial Talents Program (RC2011083), the National Natural Science Foundation of China (NSFC81373125 and NSFC81302466), and the Preventive Medicine Program funded by the Department of Health of Jiangsu Province (Y2012069).
Author Disclosure Statement
No competing financial interests exist.