
Abstract

Introduction
HIV-
Inflammatory Monocytes Are Critical Cells in the Pathogenesis of Chronic Inflammation
Monocytes are classified into three major subpopulation based on expression of CD14 and CD16 (classical, CD14++CD16−; nonclassical, CD14+CD16++; and intermediate, CD14++CD16+). Nonclassical and intermediate monocytes are considered the major inflammatory subsets. Although the specific drivers of chronic inflammation in HIV+/cART persons are not completely elucidated, current evidence suggests that increases in the inflammatory subsets of monocytes that express CDllb, indicative of activation, produce proinflammatory cytokines critical to fuelling inflammation. The precise mechanism by which these monocytes are activated and differentiated is still unclear.
Monocyte Function and Fate Depend on Metabolic Reprogramming
Cellular metabolism is an essential function of living cells and immune cells such as monocytes are no exception. 3 In monocytes, glucose is an important substrate for adenosine triphosphate (ATP) production. When activated, human monocytes dramatically increase glucose transporter-1 (Glut1) expression and change their metabolic phenotype from oxidative to glycolytic metabolism. This metabolic transition is also essential for monocyte to macrophage differentiation where Glut1 is the primary rate-limiting glucose transporter expressed on proinflammatory macrophages. 4
We observed that Glut1 is elevated on nonclassical and classical monocytes in blood from HIV-infected individuals including those receiving cART (C. Palmer and S. Crowe, unpublished data). These Glut1+ monocytes express higher TNF and IL-6 compared to Glut1− monocytes (C. Palmer and S. Crowe, unpublished data). Given these key observations we propose a model by which HIV infection both in treated and untreated individuals drives metabolic changes in monocytes and macrophages and instigates an inflammatory environment that increases the risk of age-associated comorbidities.
Glucose Metabolism in Monocytes Regulates Inflammation: A Model
Our model speculates that increased gut permeabilization in HIV-infected individuals elevates plasma levels of LPS, which triggers metabolic activation of the intermediate subset of monocytes to support the energy demands of cellular activation and inflammatory protein synthesis (Fig. 1). This premise is supported by studies demonstrating that monocyte and macrophage activation elicited by phorbol myristate acetate, or interferon (IFN)-γ and LPS, respectively, triggers Glut1 trafficking to the plasma membrane of monocytes and subsequent glucose uptake by macrophages. 4 In fact, macrophages overexpressing Glut1 exhibit a hyperinflammatory state, secreting high levels of inflammatory mediators (G-CSF, CXCL2, Il-6, CCL5, TNF, and IL-1ra) and reactive oxygen species, 4 a response that may heighten the risk of cardiovascular disease. Perhaps exploitation of drugs that modulate glucose metabolic pathways in monocytes could serve as a new class of interventions to mitigate chronic inflammation in HIV-infected people?
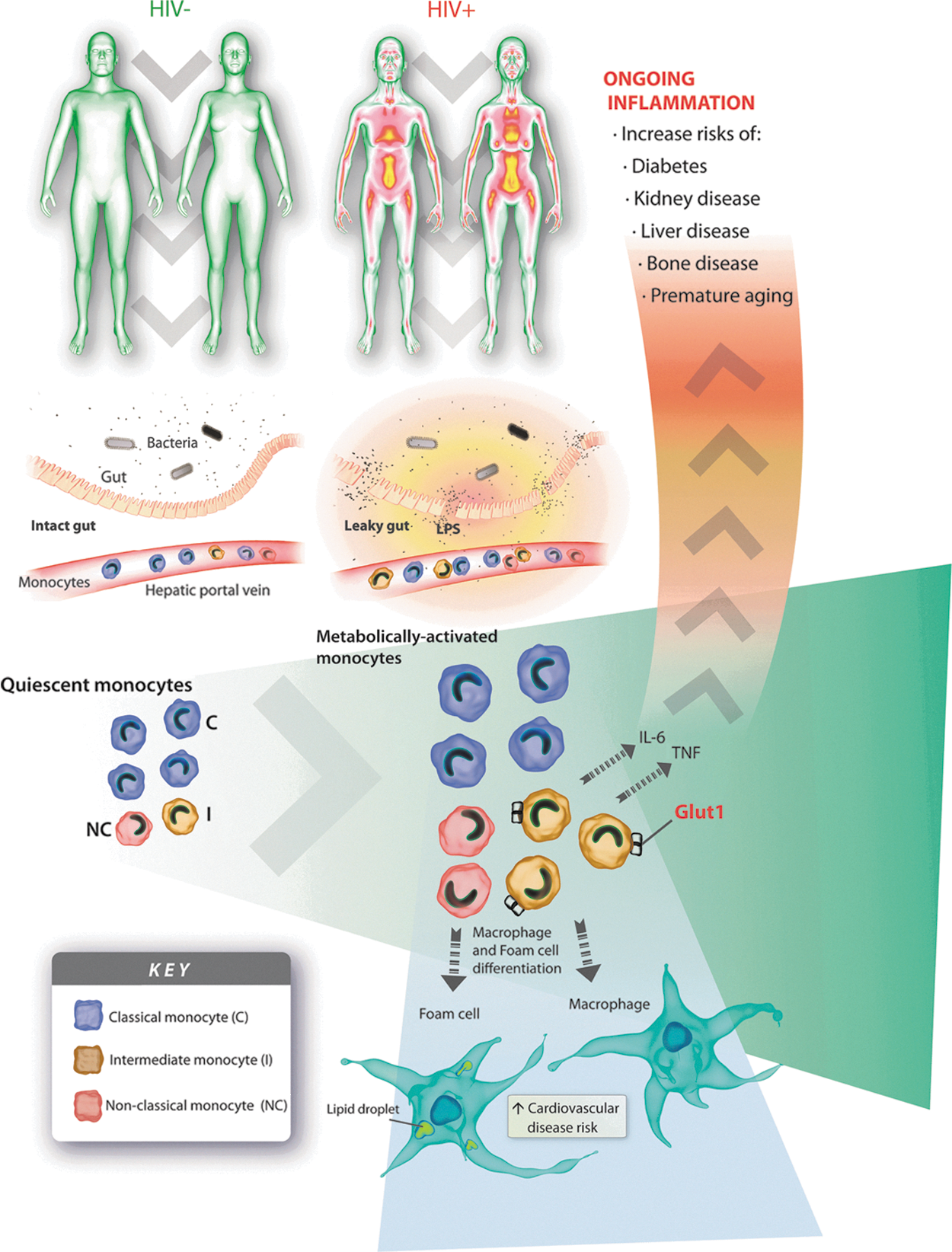
A model: how glucose metabolism in monocytes influences the development of age-related diseases in HIV+ individuals receiving antiretroviral therapy. HIV infection reduces the integrity of the gut mucosa allowing bacterial products [e.g., lipopolysaccharide (LPS) and endotoxins] to enter the gut submucosa and cross into the bloodstream. Bacterial products induce monocyte metabolic activation and a concomitant increase in glucose transporter 1 (Glut1) expression, glucose uptake, and glycolysis, predominantly within the intermediate monocyte subpopulation. Intermediate monocytes are key producers of inflammatory mediators [e.g., interleukin (IL)-6 and tumor necrosis factor (TNF)] and are a critical population implicated in chronic inflammation in HIV-infected individuals. Ongoing inflammation increases the risk of age-associated comorbidities and causes premature immune aging. Activation of monocytes also encourages CD11b expression on intermediate monocytes, prompts monocyte transition to macrophages and foam cells, and thereby increases the risk of cardiovascular disease in HIV-infected individuals. Activation of monocytes causes enzymatic shedding of CD14 (sCD14) and CD163 (sCD163) from their plasma membrane. Plasma levels of sCD14 and sCD163 are surrogate markers of immune activation and inflammation.
Footnotes
Acknowledgments
C.S.P would like to thank Mr. Wallace Wainhouse from NICE Consultants and Mr. T. Angelovich from the Burnet Institute for Medical Research for artistic support. C.S.P is a recipient of the CNIHR and ACH2 grant. S.M.C is a recipient of a National Health and Medical Research Council of Australia (NHMRC) Principal Research Fellowship. The authors gratefully acknowledge the contribution to this work of the Victorian Operational Infrastructure Support Program received by the Burnet Institute.
Author Disclosure Statement
No competing financial interests exist.